
Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer

 ,
,  , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies, Chemicals, and Reagents
2.2. Cell Culture and OSF-2 Secretion
2.3. Microarray Analysis
2.4. Clinical Data Analysis
2.5. Microscopy, Fluorescence Imaging, and Quantitation of Cells
2.6. RNA Extraction, Reverse Transcription (RT)-PCR, and Quantitative Real-Time PCR Analysis
2.7. Study Population, Tissue Preparation, and Immunohistochemistry (IHC)
2.8. Plasmids and Sequence Analyses
2.9. Protein Extraction, Immunoblot Analysis
2.10. Cellular Assays
2.11. Xenograft Experiments
2.12. Statistical Analysis
3. Results
3.1. OSF-2 Is Overexpressed in HNC Primary Tumors and Lymph Node Metastases
3.2. OSF-2 Expression Does Not Directly Contribute to Therapy Resistance
3.3. OSF-2 Is Overexpressed in the Tumor Microenvironment
3.4. HNC Patients Express Various OSF-2 Isoforms
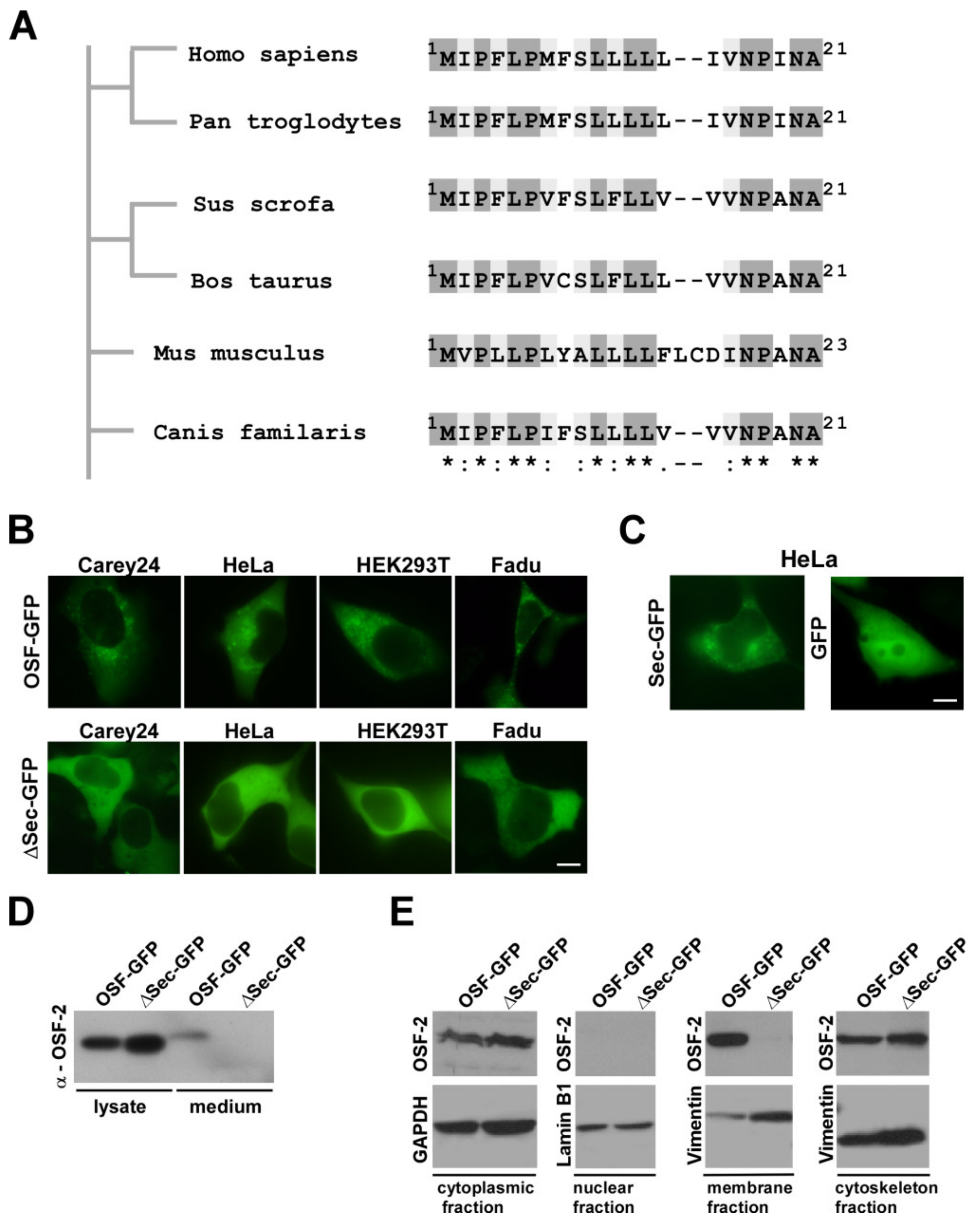
3.5. OSF-2 Isoforms of HNC Patients Contain an Evolutionary Conserved, Functional Secretion Signal
3.6. OSF-2 Does Not Affect Tumor Cell Proliferation but Is Crucial for Cell Migration and Cellular Survival under Stress Conditions
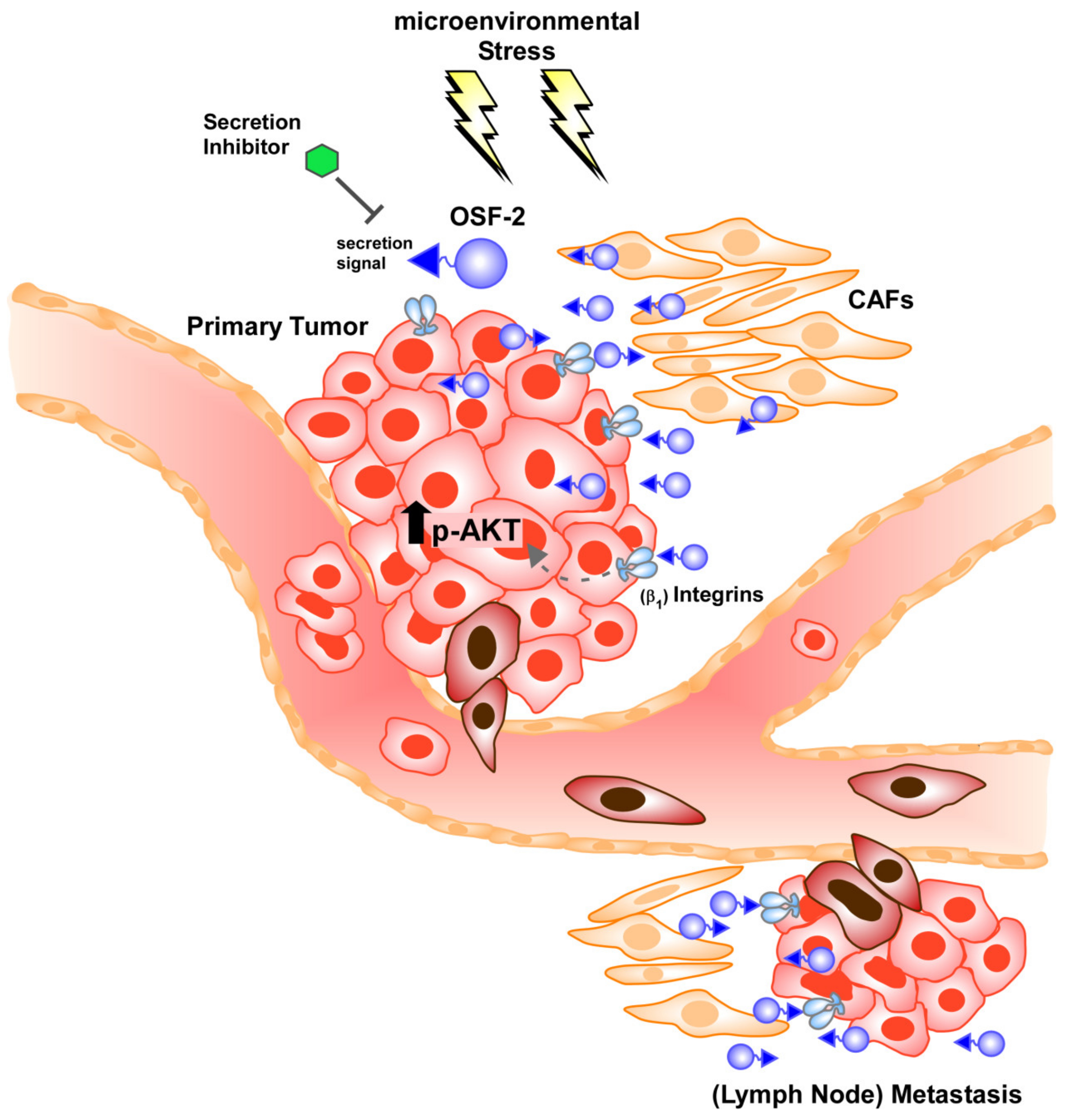
3.7. OSF-2 Promotes Cells by β1-Integrin-Induced Activation of the PI3-Kinase/Akt/PKB Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gul, D.; Habtemichael, N.; Dietrich, D.; Dietrich, J.; Gosswein, D.; Khamis, A.; Deuss, E.; Kunzel, J.; Schneider, G.; Strieth, S.; et al. Identification of cytokeratin24 as a tumor suppressor for the management of head and neck cancer. Biol. Chem. 2021; Epub ahead of print. [Google Scholar] [CrossRef]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Ferlito, A.; Shaha, A.R.; Silver, C.E.; Rinaldo, A.; Mondin, V. Incidence and sites of distant metastases from head and neck cancer. ORL J. Oto-Rhino-Laryngol. Its Relat. Spec. 2001, 63, 202–207. [Google Scholar] [CrossRef]
- Cohen, R.B. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat. Rev. 2014, 40, 567–577. [Google Scholar] [CrossRef]
- Denaro, N.; Russi, E.G.; Adamo, V.; Merlano, M.C. State-of-the-art and emerging treatment options in the management of head and neck cancer: News from 2013. Oncology 2014, 86, 212–229. [Google Scholar] [CrossRef]
- Deuss, E.; Gosswein, D.; Gul, D.; Zimmer, S.; Foersch, S.; Eger, C.S.; Limburg, I.; Stauber, R.H.; Kunzel, J. Growth Factor Receptor Expression in Oropharyngeal Squamous Cell Cancer: Her1-4 and c-Met in Conjunction with the Clinical Features and Human Papillomavirus (p16) Status. Cancers 2020, 12, 3358. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef]
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef] [Green Version]
- Kunzel, J.; Gribko, A.; Lu, Q.; Stauber, R.H.; Wunsch, D. Nanomedical detection and downstream analysis of circulating tumor cells in head and neck patients. Biol. Chem. 2019, 400, 1465–1479. [Google Scholar] [CrossRef]
- Hu, Y.; Yu, X.; Xu, G.; Liu, S. Metastasis: An early event in cancer progression. J. Cancer Res. Clin. Oncol. 2017, 143, 745–757. [Google Scholar] [CrossRef]
- Rocken, M. Early tumor dissemination, but late metastasis: Insights into tumor dormancy. J. Clin. Investig. 2010, 120, 1800–1803. [Google Scholar] [CrossRef]
- Jou, A.; Hess, J. Epidemiology and Molecular Biology of Head and Neck Cancer. Oncol. Res. Treat. 2017, 40, 328–332. [Google Scholar] [CrossRef]
- Beltz, A.; Gosswein, D.; Zimmer, S.; Limburg, I.; Wunsch, D.; Gribko, A.; Deichelbohrer, M.; Hagemann, J.; Stauber, R.H.; Kunzel, J. Staging of oropharyngeal squamous cell carcinoma of the head and neck: Prognostic features and power of the 8th edition of the UICC staging manual. Eur. J. Surg. Oncol. 2019, 45, 1046–1053. [Google Scholar] [CrossRef]
- Ginos, M.A.; Page, G.P.; Michalowicz, B.S.; Patel, K.J.; Volker, S.E.; Pambuccian, S.E.; Ondrey, F.G.; Adams, G.L.; Gaffney, P.M. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Alevizos, I.; Mahadevappa, M.; Zhang, X.; Ohyama, H.; Kohno, Y.; Posner, M.; Gallagher, G.T.; Varvares, M.; Cohen, D.; Kim, D.; et al. Oral cancer in vivo gene expression profiling assisted by laser capture microdissection and microarray analysis. Oncogene 2001, 20, 6196–6204. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.K.; Kim, H.W.; Clayman, G.L.; Coombes, M.M.; Le, B.; Lai, S.; Zhan, F.; Luna, M.A.; Hong, W.K.; Lee, J.J. Differential expression profiling of head and neck squamous carcinoma: Significance in their phenotypic and biological classification. Oncogene 2002, 21, 8206–8219. [Google Scholar] [CrossRef] [Green Version]
- Mendez, E.; Cheng, C.; Farwell, D.G.; Ricks, S.; Agoff, S.N.; Futran, N.D.; Weymuller, E.A., Jr.; Maronian, N.C.; Zhao, L.P.; Chen, C. Transcriptional expression profiles of oral squamous cell carcinomas. Cancer 2002, 95, 1482–1494. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef]
- Bockhorst, C.; Dietrich, J.; Vogt, T.J.; Stauber, R.H.; Strieth, S.; Bootz, F.; Dietrich, D.; Vos, L. The DNA methylation landscape of PD-1 (PDCD1) and adjacent lncRNA AC131097.3 in head and neck squamous cell carcinoma. Epigenomics 2021, 13, 113–127. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Egan, S.E. Progression to Metastasis of Solid Cancer. Cancers 2021, 13, 717. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.T.; Law, J.H.; Dunn, G.P.; Uppaluri, R. Emerging insights into head and neck cancer metastasis. Head Neck 2013, 35, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Hynes, N.E. Cancer: Staying together on the road to metastasis. Nature 2014, 514, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Gribko, A.; Kunzel, J.; Wunsch, D.; Lu, Q.; Nagel, S.M.; Knauer, S.K.; Stauber, R.H.; Ding, G.B. Is small smarter? Nanomaterial-based detection and elimination of circulating tumor cells: Current knowledge and perspectives. Int. J. Nanomed. 2019, 14, 4187–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.D.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Gonzalez, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Le, Q.T.; Sutphin, P.D.; Raychaudhuri, S.; Yu, S.C.; Terris, D.J.; Lin, H.S.; Lum, B.; Pinto, H.A.; Koong, A.C.; Giaccia, A.J. Identification of osteopontin as a prognostic plasma marker for head and neck squamous cell carcinomas. Clin. Cancer Res. 2003, 9, 59–67. [Google Scholar]
- Luo, S.D.; Chen, Y.J.; Liu, C.T.; Rau, K.M.; Chen, Y.C.; Tsai, H.T.; Chen, C.H.; Chiu, T.J. Osteopontin Involves Cisplatin Resistance and Poor Prognosis in Oral Squamous Cell Carcinoma. BioMed Res. Int. 2015, 2015, 508587. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Huang, Z.; Cui, D.; Ouyang, G. The Multiaspect Functions of Periostin in Tumor Progression. Adv. Exp. Med. Biol. 2019, 1132, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Watermann, C.; Pasternack, H.; Idel, C.; Ribbat-Idel, J.; Bragelmann, J.; Kuppler, P.; Offermann, A.; Jonigk, D.; Kuhnel, M.P.; Schrock, A.; et al. Recurrent HNSCC Harbor an Immunosuppressive Tumor Immune Microenvironment Suggesting Successful Tumor Immune Evasion. Clin. Cancer Res. 2021, 27, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.E.; Nor, J.E. Perivascular stem cell niche in head and neck cancer. Cancer Lett. 2013, 338, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.M.Y.; Krinsky, A.L.; Woolaver, R.A.; Wang, X.; Chen, Z.; Wang, J.H. Tumor immune microenvironment in head and neck cancers. Mol. Carcinog 2020, 59, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabieres, C. Bone marrow as a reservoir for disseminated tumor cells: A special source for liquid biopsy in cancer patients. Bonekey Rep. 2014, 3, 584. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Amizuka, N.; Takeshita, S.; Takamatsu, H.; Katsuura, M.; Ozawa, H.; Toyama, Y.; Bonewald, L.F.; Kudo, A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone Miner. Res. 1999, 14, 1239–1249. [Google Scholar] [CrossRef]
- Takeshita, S.; Kikuno, R.; Tezuka, K.; Amann, E. Osteoblast-specific factor 2: Cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem. J. 1993, 294 Pt 1, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Elkins, T.; Hortsch, M.; Bieber, A.J.; Snow, P.M.; Goodman, C.S. Drosophila fasciclin I is a novel homophilic adhesion molecule that along with fasciclin III can mediate cell sorting. J. Cell Biol. 1990, 110, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Zhou, W.; Wang, Z.; Wang, Y.; Liu, M.; Zhang, G.; Guo, X.; Kang, X. Periostin: An Emerging Molecule With a Potential Role in Spinal Degenerative Diseases. Front. Med. 2021, 8, 694800. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, S.J.; Lee, B.H.; Park, R.W.; Kim, K.S.; Kim, I.S. Identification of motifs for cell adhesion within the repeated domains of transforming growth factor-beta-induced gene, betaig-h3. J. Biol. Chem. 2000, 275, 30907–30915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kzhyshkowska, J.; Gratchev, A.; Martens, J.H.; Pervushina, O.; Mamidi, S.; Johansson, S.; Schledzewski, K.; Hansen, B.; He, X.; Tang, J.; et al. Stabilin-1 localizes to endosomes and the trans-Golgi network in human macrophages and interacts with GGA adaptors. J. Leukoc. Biol. 2004, 76, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Bao, S.; Ouyang, G. The multifaceted role of periostin in tumorigenesis. Cell. Mol. Life Sci. 2009, 66, 2219–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Ouyang, G.; Bai, X.; Huang, Z.; Ma, C.; Liu, M.; Shao, R.; Anderson, R.M.; Rich, J.N.; Wang, X.F. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 2004, 5, 329–339. [Google Scholar] [CrossRef]
- Baril, P.; Gangeswaran, R.; Mahon, P.C.; Caulee, K.; Kocher, H.M.; Harada, T.; Zhu, M.; Kalthoff, H.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: Role of the beta4 integrin and the PI3k pathway. Oncogene 2007, 26, 2082–2094. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Wu, K.; Wang, X.; Zhang, J.; Wang, L.; Jiang, Y.; Zhu, X.; Chen, W.; Yan, M. Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.; Zhou, C.; Wang, S.; Deng, H.; Shen, Z. Tumor suppression effect of targeting periostin with siRNA in a nude mouse model of human laryngeal squamous cell carcinoma. J. Clin. Lab. Anal. 2019, 33, e22622. [Google Scholar] [CrossRef] [Green Version]
- Siemer, S.; Fauth, T.; Scholz, P.; Al-Zamel, Y.; Khamis, A.; Gul, D.; Freudelsperger, L.; Wollenberg, B.; Becker, S.; Stauber, R.H.; et al. Profiling Cisplatin Resistance in Head and Neck Cancer: A Critical Role of the VRAC Ion Channel for Chemoresistance. Cancers 2021, 13, 4831. [Google Scholar] [CrossRef]
- Welkoborsky, H.J.; Jacob, R.; Riazimand, S.H.; Bernauer, H.S.; Mann, W.J. Molecular biologic characteristics of seven new cell lines of squamous cell carcinomas of the head and neck and comparison to fresh tumor tissue. Oncology 2003, 65, 60–71. [Google Scholar] [CrossRef]
- Gribko, A.; Stiefel, J.; Liebetanz, L.; Nagel, S.M.; Künzel, J.; Wandrey, M.; Hagemann, J.; Stauber, R.H.; Freese, C.; Gül, D. IsoMAG—An Automated System for the Immunomagnetic Isolation of Squamous Cell Carcinoma-Derived Circulating Tumor Cells. Diagnostics 2021, 11, 2040. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; De Meyer, T.; Jeschke, J.; Van Criekinge, W. MEXPRESS: Visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 2015, 16, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingerdissen, H.M.; Torcivia-Rodriguez, J.; Hu, Y.; Chang, T.C.; Mazumder, R.; Kahsay, R. BioMuta and BioXpress: Mutation and expression knowledgebases for cancer biomarker discovery. Nucleic. Acids Res. 2018, 46, D1128–D1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habtemichael, N.; Wunsch, D.; Bier, C.; Tillmann, S.; Unruhe, B.; Frauenknecht, K.; Heinrich, U.R.; Mann, W.J.; Stauber, R.H.; Knauer, S.K. Cloning and functional characterization of the guinea pig apoptosis inhibitor protein Survivin. Gene 2010, 469, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bier, C.; Knauer, S.K.; Klapthor, A.; Schweitzer, A.; Rekik, A.; Kramer, O.H.; Marschalek, R.; Stauber, R.H. Cell-based Analysis of Structure-Function Activity of Threonine Aspartase 1. J. Biol. Chem. 2011, 286, 3007–3017. [Google Scholar] [CrossRef] [Green Version]
- Knauer, S.K.; Heinrich, U.R.; Bier, C.; Habtemichael, N.; Docter, D.; Helling, K.; Mann, W.J.; Stauber, R.H. An otoprotective role for the apoptosis inhibitor protein survivin. Cell Death Dis. 2010, 1, e51. [Google Scholar] [CrossRef] [Green Version]
- Engels, K.; Knauer, S.K.; Loibl, S.; Fetz, V.; Harter, P.; Schweitzer, A.; Fisseler-Eckhoff, A.; Kommoss, F.; Hanker, L.; Nekljudova, V.; et al. NO signaling confers cytoprotectivity through the survivin network in ovarian carcinomas. Cancer Res. 2008, 68, 5159–5166. [Google Scholar] [CrossRef] [Green Version]
- Fetz, V.; Bier, C.; Habtemichael, N.; Schuon, R.; Schweitzer, A.; Kunkel, M.; Engels, K.; Kovacs, A.F.; Schneider, S.; Mann, W.; et al. Inducible NO synthase confers chemoresistance in head and neck cancer by modulating survivin. Int. J. Cancer 2009, 124, 2033–2041. [Google Scholar] [CrossRef]
- Trothe, J.; Ritzmann, D.; Lang, V.; Scholz, P.; Pul, U.; Kaufmann, R.; Buerger, C.; Ertongur-Fauth, T. Hypotonic stress response of human keratinocytes involves LRRC8A as component of volume-regulated anion channels. Exp. Dermatol. 2018, 27, 1352–1360. [Google Scholar] [CrossRef]
- Stauber, R.H.; Rabenhorst, U.; Rekik, A.; Engels, K.; Bier, C.; Knauer, S.K. Nucleocytoplasmic shuttling and the biological activity of mouse survivin are regulated by an active nuclear export signal. Traffic 2006, 7, 1461–1472. [Google Scholar] [CrossRef]
- Stauber, R.H.; Knauer, S.K.; Habtemichael, N.; Bier, C.; Unruhe, B.; Weisheit, S.; Spange, S.; Nonnenmacher, F.; Fetz, V.; Ginter, T.; et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget 2012, 3, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Stauber, R.H.; Bier, C.; Knauer, S.K. Targeting Taspase1 for cancer therapy—letter. Cancer Res. 2012, 72, 2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef] [Green Version]
- Price, K.A.; Cohen, E.E. Current treatment options for metastatic head and neck cancer. Curr. Treat. Options Oncol. 2012, 13, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Oosting, S.F.; Haddad, R.I. Best Practice in Systemic Therapy for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Siemer, S.; Bauer, T.A.; Scholz, P.; Breder, C.; Fenaroli, F.; Harms, G.; Dietrich, D.; Dietrich, J.; Rosenauer, C.; Barz, M.; et al. Targeting Cancer Chemotherapy Resistance by Precision Medicine-Driven Nanoparticle-Formulated Cisplatin. ACS Nano 2021, 15, 18541–18556. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jin, P.; Liu, W. Periostin Contributes to Cisplatin Resistance in Human Non-Small Cell Lung Cancer A549 Cells via Activation of Stat3 and Akt and Upregulation of Survivin. Cell. Physiol. Biochem. 2016, 38, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Engels, K.; du Bois, A.; Harter, P.; Fisseler-Eckhoff, A.; Kommoss, F.; Stauber, R.; Kaufmann, M.; Nekljudova, V.; Loibl, S. VEGF-A and i-NOS Expression are Prognostic Factors in Serous Epithelial Ovarian Carcinomas after Complete Surgical Resection. J. Clin. Pathol. 2009, 62, 448–454. [Google Scholar] [CrossRef]
- Engels, K.; Knauer, S.K.; Metzler, D.; Simf, C.; Struschka, O.; Bier, C.; Mann, W.; Kovacs, A.F.; Stauber, R.H. Dynamic intracellular survivin in oral squamous cell carcinoma: Underlying molecular mechanism and potential as an early prognostic marker. J. Pathol. 2007, 211, 532–540. [Google Scholar] [CrossRef]
- Litvin, J.; Selim, A.H.; Montgomery, M.O.; Lehmann, K.; Rico, M.C.; Devlin, H.; Bednarik, D.P.; Safadi, F.F. Expression and function of periostin-isoforms in bone. J. Cell. Biochem. 2004, 92, 1044–1061. [Google Scholar] [CrossRef]
- Khurshid, Z.; Mali, M.; Adanir, N.; Zafar, M.S.; Khan, R.S.; Latif, M. Periostin: Immunomodulatory Effects on Oral Diseases. Eur. J. Dent. 2020, 14, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Kii, I. Periostin Functions as a Scaffold for Assembly of Extracellular Proteins. Adv. Exp. Med. Biol. 2019, 1132, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Kunita, A.; Iwata, C.; Komura, D.; Nishiyama, T.; Shimazu, K.; Takeshita, K.; Shibahara, J.; Kii, I.; Morishita, Y.; et al. The niche component periostin is produced by cancer-associated fibroblasts, supporting growth of gastric cancer through ERK activation. Am. J. Pathol. 2014, 184, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Iizuka, S.; Yoshida, M.; Nguyen, P.T.; Siriwardena, S.B.; Tsunematsu, T.; Ohbayashi, M.; Ando, T.; Hatakeyama, D.; Shibata, T.; et al. Periostin directly and indirectly promotes tumor lymphangiogenesis of head and neck cancer. PLoS ONE 2012, 7, e44488. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, K.; Yoon, J.W.; Choi, S.H.; Sheen, Y.H.; Han, M.; Ono, J.; Izuhara, K.; Baek, H. Serum levels of periostin and exercise-induced bronchoconstriction in asthmatic children. World Allergy Organ. J. 2019, 12, 100004. [Google Scholar] [CrossRef] [Green Version]
- Kanno, A.; Satoh, K.; Masamune, A.; Hirota, M.; Kimura, K.; Umino, J.; Hamada, S.; Satoh, A.; Egawa, S.; Motoi, F.; et al. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int. J. Cancer 2008, 122, 2707–2718. [Google Scholar] [CrossRef]
- Ma, H.; Wang, J.; Zhao, X.; Wu, T.; Huang, Z.; Chen, D.; Liu, Y.; Ouyang, G. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep. 2020, 30, 793–806.e6. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.; Hah, Y.S.; Ryu, S.; Cheon, S.Y.; Cho, H.Y.; Kim, J.P.; Won, S.J.; Lee, J.S.; Hwa, J.S.; Seo, J.H.; et al. Periostin Plays a Key Role in Radioresistance of Head and Neck Cancer Cells Via Epithelial-to-Mesenchymal Transition. Anticancer Res. 2020, 40, 2627–2635. [Google Scholar] [CrossRef]
- Martinez, I.; Wang, J.; Hobson, K.F.; Ferris, R.L.; Khan, S.A. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur. J. Cancer 2007, 43, 415–432. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Yu, C.Y.; Dai, M.; Tam, C.; Loda, M.; Auclair, D.; Chen, L.B.; Elias, A. Elevated serum periostin levels in patients with bone metastases from breast but not lung cancer. Breast Cancer Res. Treat. 2003, 77, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Buchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Wang, F.; Zhang, W.; Li, H.F.; Xu, J.; Tong, X.W. Periostin Secreted by Carcinoma-Associated Fibroblasts Promotes Ovarian Cancer Cell Platinum Resistance Through the PI3K/Akt Signaling Pathway. Technol. Cancer Res. Treat. 2020, 19, 1533033820977535. [Google Scholar] [CrossRef]
- Sung, P.L.; Jan, Y.H.; Lin, S.C.; Huang, C.C.; Lin, H.; Wen, K.C.; Chao, K.C.; Lai, C.R.; Wang, P.H.; Chuang, C.M.; et al. Periostin in tumor microenvironment is associated with poor prognosis and platinum resistance in epithelial ovarian carcinoma. Oncotarget 2016, 7, 4036–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterzynska, K.; Kazmierczak, D.; Klejewski, A.; Swierczewska, M.; Wojtowicz, K.; Nowacka, M.; Brazert, J.; Nowicki, M.; Januchowski, R. Expression of Osteoblast-Specific Factor 2 (OSF-2, Periostin) Is Associated with Drug Resistance in Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Wang, Z.; Zhang, L.; Feng, Y.; Lv, J.; Wu, Z.; Yang, R.; Wu, T.; Li, J.; Zhou, R.; et al. Periostin promotes the proliferation and metastasis of osteosarcoma by increasing cell survival and activates the PI3K/Akt pathway. Cancer Cell Int. 2022, 22, 34. [Google Scholar] [CrossRef]
- Xouri, G.; Christian, S. Origin and function of tumor stroma fibroblasts. Semin. Cell Dev. Biol. 2010, 21, 40–46. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.U.; Yun, J.S.; Lee, I.H.; Heo, S.C.; Shin, S.H.; Jeon, E.S.; Choi, Y.J.; Suh, D.S.; Yoon, M.S.; Kim, J.H. Lysophosphatidic acid-induced expression of periostin in stromal cells: Prognoistic relevance of periostin expression in epithelial ovarian cancer. Int. J. Cancer 2011, 128, 332–342. [Google Scholar] [CrossRef]
- Shao, R.; Bao, S.; Bai, X.; Blanchette, C.; Anderson, R.M.; Dang, T.; Gishizky, M.L.; Marks, J.R.; Wang, X.F. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol. Cell. Biol. 2004, 24, 3992–4003. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.J.; Yoshioka, N.; Tambe, Y.; Kushima, R.; Okada, Y.; Inoue, H. Periostin is down-regulated in high grade human bladder cancers and suppresses in vitro cell invasiveness and in vivo metastasis of cancer cells. Int. J. Cancer 2005, 117, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ismail, R.S.; Baldwin, R.L.; Fang, J.; Browning, D.; Karlan, B.Y.; Gasson, J.C.; Chang, D.D. Differential gene expression between normal and tumor-derived ovarian epithelial cells. Cancer Res. 2000, 60, 6744–6749. [Google Scholar] [PubMed]
- Gonzalez, H.E.; Gujrati, M.; Frederick, M.; Henderson, Y.; Arumugam, J.; Spring, P.W.; Mitsudo, K.; Kim, H.W.; Clayman, G.L. Identification of 9 genes differentially expressed in head and neck squamous cell carcinoma. Arch. Otolaryngol.-Head Neck Surg. 2003, 129, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, N.; Fuji, S.; Shimakage, M.; Kodama, K.; Hakura, A.; Yutsudo, M.; Inoue, H.; Nojima, H. Suppression of anchorage-independent growth of human cancer cell lines by the TRIF52/periostin/OSF-2 gene. Exp. Cell Res. 2002, 279, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.T.; Dai, M.; Chen, L.B. Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogenesis 2005, 26, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Dai, M.; Auclair, D.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y.; Chen, L.B. Serum level of the periostin, a homologue of an insect cell adhesion molecule, as a prognostic marker in nonsmall cell lung carcinomas. Cancer 2001, 92, 843–848. [Google Scholar] [CrossRef]
- Sasaki, H.; Lo, K.M.; Chen, L.B.; Auclair, D.; Nakashima, Y.; Moriyama, S.; Fukai, I.; Tam, C.; Loda, M.; Fujii, Y. Expression of Periostin, homologous with an insect cell adhesion molecule, as a prognostic marker in non-small cell lung cancers. Jpn. J. Cancer Res. 2001, 92, 869–873. [Google Scholar] [CrossRef]
- Shao, R.; Guo, X. Human microvascular endothelial cells immortalized with human telomerase catalytic protein: A model for the study of in vitro angiogenesis. Biochem. Biophys. Res. Commun. 2004, 321, 788–794. [Google Scholar] [CrossRef]
- Vardaki, I.; Ceder, S.; Rutishauser, D.; Baltatzis, G.; Foukakis, T.; Panaretakis, T. Periostin is identified as a putative metastatic marker in breast cancer-derived exosomes. Oncotarget 2016, 7, 74966–74978. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, M.; Schinelli, S. Integrins and Exosomes, a Dangerous Liaison in Cancer Progression. Cancers 2017, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, I.S.; Park, S.A.; Kim, Y.; Lee, J.E.; Noh, D.Y.; Kim, K.T.; Ryu, S.H.; Suh, P.G. Periostin-binding DNA aptamer inhibits breast cancer growth and metastasis. Mol. Ther. 2013, 21, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, K.K.; Kamolhan, T.; Soni, A.; Thongchot, S.; Mitrpant, C.; Thuwajit, C.; Thuwajit, P. Development of an engineered peptide antagonist against periostin to overcome doxorubicin resistance in breast cancer. BMC Cancer 2021, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Kyutoku, M.; Taniyama, Y.; Katsuragi, N.; Shimizu, H.; Kunugiza, Y.; Iekushi, K.; Koibuchi, N.; Sanada, F.; Oshita, Y.; Morishita, R. Role of periostin in cancer progression and metastasis: Inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int. J. Mol. Med. 2011, 28, 181–186. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gül, D.; Schweitzer, A.; Khamis, A.; Knauer, S.K.; Ding, G.-B.; Freudelsperger, L.; Karampinis, I.; Strieth, S.; Hagemann, J.; Stauber, R.H. Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer. Cancers 2022, 14, 2337. https://doi.org/10.3390/cancers14092337
Gül D, Schweitzer A, Khamis A, Knauer SK, Ding G-B, Freudelsperger L, Karampinis I, Strieth S, Hagemann J, Stauber RH. Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer. Cancers. 2022; 14(9):2337. https://doi.org/10.3390/cancers14092337
Chicago/Turabian StyleGül, Désirée, Andrea Schweitzer, Aya Khamis, Shirley K. Knauer, Guo-Bin Ding, Laura Freudelsperger, Ioannis Karampinis, Sebastian Strieth, Jan Hagemann, and Roland H. Stauber. 2022. "Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer" Cancers 14, no. 9: 2337. https://doi.org/10.3390/cancers14092337