
Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population

 , , , , , , , , add
Show full author list
, , , , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The EU-RHAB Registry
2.2. Consensus Multimodal Therapy
2.3. Diagnostic Measures
2.4. Toxicity
2.5. Tumor Tissue Collection and Genetic Analyses
2.6. Statistical Analyses
3. Results
3.1. Patient Characteristics
3.2. Clinical Factors Associated with Outcome
3.3. Germline Mutation of SMARCB1 an Important Risk Factor
3.4. Distribution of ATRT Subgroups in This Cohort
3.5. Extent of Surgery
3.6. Systemic, Intrathecal and Maintenance Chemotherapy
3.7. Radiotherapy
3.8. Survival
3.9. Toxicity of Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Statistics Reports for the Germany. Available online: http://www.Kinderkrebsregister.De/dkkr/ergebnisse/jahresberichte/jahresbericht-2019 (accessed on 1 December 2021).
- Brennan, B.; Stiller, C.; Bourdeaut, F. Extracranial rhabdoid tumours: What we have learned so far and future directions. Lancet Oncol. 2013, 14, e329–e336. [Google Scholar] [CrossRef]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: A report from the children’s oncology group trial acns0333. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, M.C.; Hasselblatt, M.; Nemes, K.; Bens, S.; Steinbügl, M.; Johann, P.D.; Kerl, K.; Hauser, P.; Quiroga, E.; Solano-Paez, P.; et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro-Oncology 2020, 22, 1006–1017. [Google Scholar] [CrossRef]
- Nemes, K.; Bens, S.; Kachanov, D.; Teleshova, M.; Hauser, P.; Simon, T.; Tippelt, S.; Woessmann, W.; Beck, O.; Flotho, C.; et al. Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (emrt/rtk). Eur. J. Cancer 2021, 142, 112–122. [Google Scholar] [CrossRef]
- Brennan, B.; De Salvo, G.L.; Orbach, D.; De Paoli, A.; Kelsey, A.; Mudry, P.; Francotte, N.; Van Noesel, M.; Bisogno, G.; Casanova, M.; et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the european paediatric soft tissue sarcoma study group non-rhabdomyosarcoma soft tissue sarcoma 2005 study-epssg nrsts 2005. Eur. J. Cancer 2016, 60, 69–82. [Google Scholar] [CrossRef]
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental pharmacology—Drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 2003, 349, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Schmal, Z.; Isermann, A.; Hladik, D.; von Toerne, C.; Tapio, S.; Rübe, C.E. DNA damage accumulation during fractionated low-dose radiation compromises hippocampal neurogenesis. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2019, 137, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bojaxhiu, B.; Ahlhelm, F.; Walser, M.; Placidi, L.; Kliebsch, U.; Mikroutsikos, L.; Morach, P.; Bolsi, A.; Lomax, T.; Pica, A.; et al. Radiation necrosis and white matter lesions in pediatric patients with brain tumors treated with pencil beam scanning proton therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zając-Spychała, O.; Pawlak, M.A.; Karmelita-Katulska, K.; Pilarczyk, J.; Derwich, K.; Wachowiak, J. Long-term brain structural magnetic resonance imaging and cognitive functioning in children treated for acute lymphoblastic leukemia with high-dose methotrexate chemotherapy alone or combined with cns radiotherapy at reduced total dose to 12 gy. Neuroradiology 2017, 59, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Hertzberg, H.; Huk, W.J.; Ueberall, M.A.; Langer, T.; Meier, W.; Dopfer, R.; Skalej, M.; Lackner, H.; Bode, U.; Janssen, G.; et al. Cns late effects after all therapy in childhood. Part i: Neuroradiological findings in long-term survivors of childhood all--an evaluation of the interferences between morphology and neuropsychological performance. The german late effects working group. Med. Pediatr. Oncol. 1997, 28, 387–400. [Google Scholar] [CrossRef]
- Marks, J.E.; Baglan, R.J.; Prassad, S.C.; Blank, W.F. Cerebral radionecrosis: Incidence and risk in relation to dose, time, fractionation and volume. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 243–252. [Google Scholar] [CrossRef]
- Nemes, K.; Clément, N.; Kachanov, D.; Bens, S.; Hasselblatt, M.; Timmermann, B.; Schneppenheim, R.; Gerss, J.; Siebert, R.; Furtwängler, R.; et al. The extraordinary challenge of treating patients with congenital rhabdoid tumors-a collaborative european effort. Pediatr. Blood Cancer 2018, 65, e26999. [Google Scholar] [CrossRef] [PubMed]
- Seeringer, A.; Bartelheim, K.; Kerl, K.; Hasselblatt, M.; Leuschner, I.; Rutkowski, S.; Timmermann, B.; Kortmann, R.-D.; Koscielniak, E.; Schneppenheim, R.; et al. Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors—Experience of the eu-rhab registry. Klin. Pädiatr. 2014, 226, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Judkins, A.R.; Mauger, J.; Ht, A.; Rorke, L.B.; Biegel, J.A. Immunohistochemical analysis of hsnf5/ini1 in pediatric cns neoplasms. Am. J. Surg. Pathol. 2004, 28, 644–650. [Google Scholar] [CrossRef]
- Kordes, U.; Bartelheim, K.; Modena, P.; Massimino, M.; Biassoni, V.; Reinhard, H.; Hasselblatt, M.; Schneppenheim, R.; Fruhwald, M.C. Favorable outcome of patients affected by rhabdoid tumors due to rhabdoid tumor predisposition syndrome (rtps). Pediatr. Blood Cancer 2014, 61, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Gesk, S.; Frühwald, M.C.; Leuschner, I.; Hasselblatt, M.; Jeibmann, A.; Oyen, F.; Peters, O.; Pietsch, T.; Siebert, R.; et al. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant tumor. Genes Chrom. Cancer 2010, 49, 176–181. [Google Scholar] [CrossRef]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors—A reinvestigation and current consensus. Nat. Med. 2020, 22, 613–624. [Google Scholar]
- Thomas, C.; Knerlich-Lukoschus, F.; Reinhard, H.; Johann, P.D.; Sturm, D.; Sahm, F.; Bens, S.; Vogt, J.; Nemes, K.; Oyen, F.; et al. Two molecularly distinct atypical teratoid/rhabdoid tumors (or tumor components) occurring in an infant with rhabdoid tumor predisposition syndrome 1. Acta Neuropathol. 2019, 137, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Schneppenheim, R.; Fruhwald, M.C.; Gesk, S.; Hasselblatt, M.; Jeibmann, A.; Kordes, U.; Kreuz, M.; Leuschner, I.; Martin Subero, J.I.; Obser, T.; et al. Germline nonsense mutation and somatic inactivation of smarca4/brg1 in a family with rhabdoid tumor predisposition syndrome. Am. J. Hum. Genet. 2010, 86, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, G.E.; Breslow, N.E.; Dome, J.; Guthrie, K.A.; Norkool, P.; Li, S.; Thomas, P.R.; Perlman, E.; Beckwith, J.B.; D’Angio, G.J.; et al. Rhabdoid tumor of the kidney in the national wilms’ tumor study: Age at diagnosis as a prognostic factor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 7641–7645. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Qaddoumi, I.; Rodríguez-Galindo, C.; Nassan, A.A.; Ghandour, K.; Al-Hussaini, M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr. Blood Cancer 2010, 54, 35–40. [Google Scholar] [CrossRef]
- Arndt, C.; Hawkins, D.; Anderson, J.R.; Breitfeld, P.; Womer, R.; Meyer, W. Age is a risk factor for chemotherapy-induced hepatopathy with vincristine, dactinomycin, and cyclophosphamide. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Jagt, C.T.; Zuckermann, M.; Ten Kate, F.; Taminiau, J.A.; Dijkgraaf, M.G.; Heij, H.; De Kraker, J.; Verschuur, A.C. Veno-occlusive disease as a complication of preoperative chemotherapy for wilms tumor: A clinico-pathological analysis. Pediatr. Blood Cancer 2009, 53, 1211–1215. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered b7-h3-targeted car t cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | |
|---|---|
| Median age [months] | 3 (0–6) |
| Origin [n = 100] | |
| Germany/Austria/Switzerland | 74 |
| Other countries | 26 |
| Sex [n = 100] | |
| Female | 49 |
| Male | 51 |
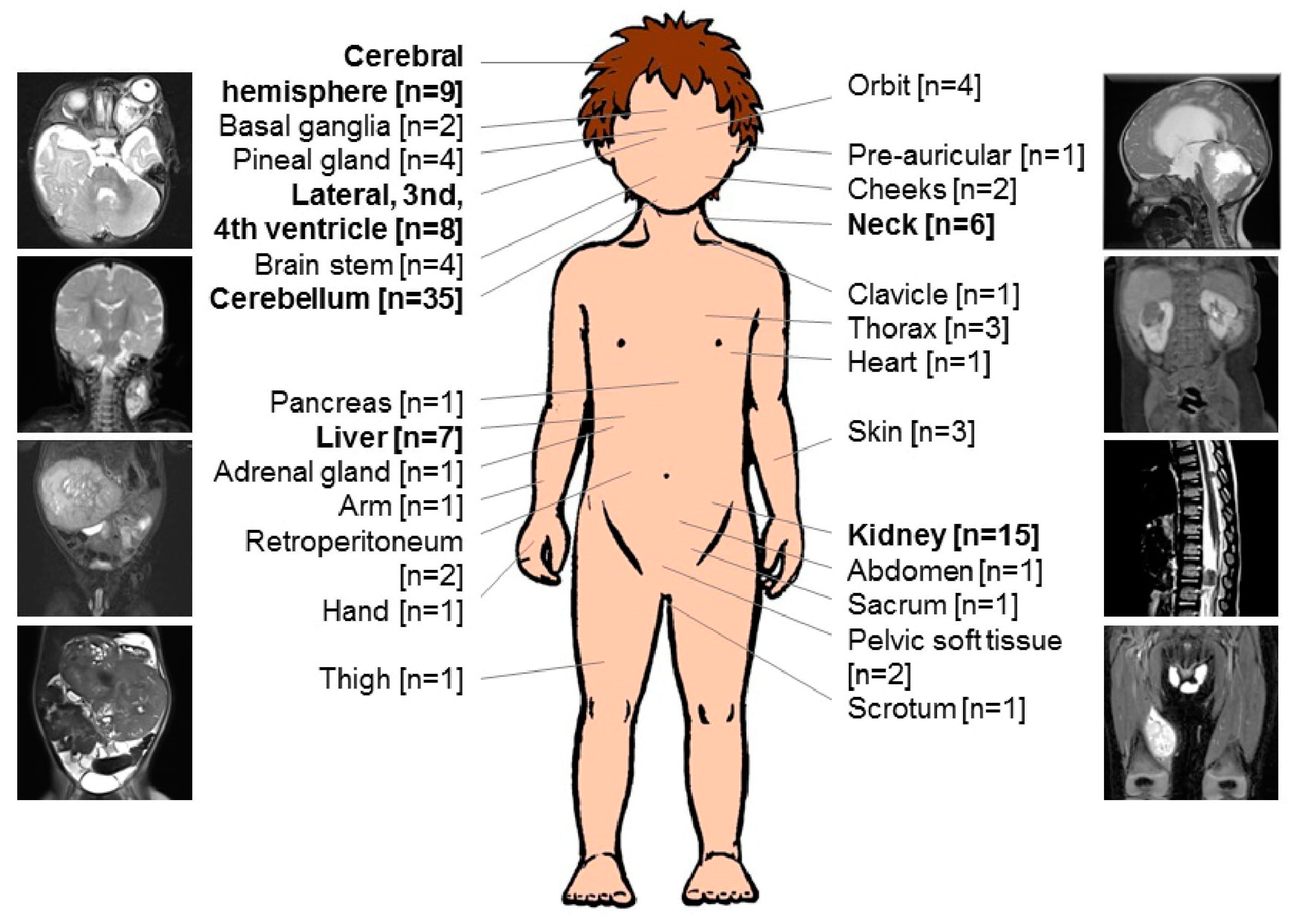
| Localization [n = 117] * | |
| Intracranial MRT (ATRT) | 62 |
| Cerebellum | 35 |
| Cerebral hemisphere | 9 |
| Lateral, 3rd, 4th ventricle | 8 |
| Pineal gland | 4 |
| Brain stem | 4 |
| Basal ganglia | 2 |
| Extracranial MRT | 55 |
| Kidney | 15 |
| Liver | 7 |
| Neck | 6 |
| Orbit | 4 |
| Thorax | 3 |
| Skin | 3 |
| Retroperitoneum | 2 |
| Pelvic soft tissue | 2 |
| Cheeks | 2 |
| Pre-auricular | 1 |
| Clavicle | 1 |
| Heart | 1 |
| Pancreas | 1 |
| Abdomen | 1 |
| Adrenal gland | 1 |
| Arm | 1 |
| Hand | 1 |
| Thigh | 1 |
| Sacrum | 1 |
| Scrotum | 1 |
| Metastasis [n = 97] ** | |
| M0, LN− | 49 |
| M0, LN+ | 5 |
| M+ | 26 |
| Synchronous tumor | 17 |
| Total [n] | |
|---|---|
| Gross total resection [n = 100] | |
| Yes | 32 |
| No | 65 |
| Any radiotherapy [n = 100] | |
| Yes | 24 |
| No | 76 |
| High dose chemotherapy [n = 93] * | |
| Yes | 16 |
| No | 77 |
| Maintenance therapy [n = 93] * | |
| Yes | 18 |
| No | 75 |
| Complete remission (of all sites involved) [n = 100] | |
| Yes | 34 |
| After surgery | 5 |
| + chemotherapy | 25 |
| + radiotherapy | 4 |
| No | 66 |
| Progression [n = 100] | |
| No | 22 |
| PD on CT ** | 56 |
| PD after CT *** | 22 |
| SAE [n = 17] | 17 |
| VOD | 12 |
| Encephalomalacia | 1 |
| Sinus vein thrombosis | 1 |
| Shunt failure | 1 |
| Sinus tachycardia | 1 |
| AML | 1 |
| Present status [n = 100] | |
| Complete remission | 18 |
| Stable disease | 1 |
| Progressive disease | 3 |
| Death | 78 |
| Prognostic Factors | Univariate Analysis n = 93 | Multivariate Analysis n = 81 | |
|---|---|---|---|
| p-Value | RR (95% CI) | p-Value | |
| Gender M versus F | 0.03 | 2.1 (1.2–3.6) | 0.007 |
| M+ versus M0 | 0.0006 | ||
| SYN yes versus no | 0.045 | ||
| M+/SYN versus M0, LN−/M0, LN+ | 3.3 (1.8–6) | 0.0001 | |
| GLM yes versus no | 0.0009 | 2 (1.1–3.6) | 0.02 |
| MYC versus TYR * | 0.0005 | ||
| GTR yes versus no | 0.24 | ||
| HDCT yes versus no | 0.23 | ||
| RTx yes versus no | 0.35 | ||
| MT yes versus no | 0.075 | 0.3 (0.1–0.8) | 0.02 |
| CR yes versus no | 0.0003 | ||
| Patient | Age at Diagnosis (Months) | Primary Tumor Lokalisation | VOD | Current Status(Months) |
|---|---|---|---|---|
| 1 | 4 | Cerebellum + neck (SYN, M0, LN+, GTR cerebellum, GTR neck, CR) | After the third course of VAC, recovered completely | LR (6)–DOD (26) |
| 2 | 0 | Pineal gland + pelvic soft tissue (SYN, M0, GTR pineal gland, not operated pelvis, PD) | After the third course of VAC, recovered completely | PD (2)–DOD (5) |
| 3 | 4 | RTK (M0, GTR, CR) | After the third course of VCA, recovered completely | CR (108) |
| 4 | 6 | Cerebellum (M0, STR, PD) | After the third course of VCA recovered completely | PD (6)–DOD (7) |
| 5 | 3 | Pineal gland + kidney (SYN, M0, LN−, biopsy pineal gland, GTR kidney, PD) | After the third course of VA, recovered completely | PD (6)–DOD (7) |
| 6 | 0 | Cerebellum (M0, STR, SD) | After the third course of VCA, recovered completely | PD (8)–DOD (13) |
| 7 | 5 | Cerebellum (M0, STR, PD) | After the third course of VCA, recovered completely | PD (8)–DOD (11) |
| 8 | 2 | Basal ganglia (M0, biopsy, PD) | After the third course of VCA, recovered completely | PD (5)–DOD (8) |
| 9 | 4 | Cerebellum (M0, PR, SD) | After the first course of HDCT, recovered completely | CR (43) |
| 10 | 1 | Cerebellum (M1, GTR, CR) | After the first course of HDCT | CR (5)–DOT (7) |
| 11 | 0 | Scrotum (M0, LN−, GTR, CR) | After the third course of VAC | CR (1)–DOT (1) |
| 12 | 1 | Heart + Tectum mesencephali (SYN, M0, LN−, biopsy heart, PD) | After achieving one course of VAC | PD (0)–DOT (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemes, K.; Johann, P.D.; Steinbügl, M.; Gruhle, M.; Bens, S.; Kachanov, D.; Teleshova, M.; Hauser, P.; Simon, T.; Tippelt, S.; et al. Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population. Cancers 2022, 14, 2185. https://doi.org/10.3390/cancers14092185
Nemes K, Johann PD, Steinbügl M, Gruhle M, Bens S, Kachanov D, Teleshova M, Hauser P, Simon T, Tippelt S, et al. Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population. Cancers. 2022; 14(9):2185. https://doi.org/10.3390/cancers14092185
Chicago/Turabian StyleNemes, Karolina, Pascal D. Johann, Mona Steinbügl, Miriam Gruhle, Susanne Bens, Denis Kachanov, Margarita Teleshova, Peter Hauser, Thorsten Simon, Stephan Tippelt, and et al. 2022. "Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population" Cancers 14, no. 9: 2185. https://doi.org/10.3390/cancers14092185