
Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Immunohistochemical Analysis and Quantification of Protein Expression
2.2. Cell Culture and Lentiviral Transduction
2.3. MTT Cell Proliferation Assay
2.4. Migration and Invasion Assays
2.5. RNA isolation and Quantitative Real-Time PCR (qRT-PCR)
2.6. Isolation and TaqMan PCR Analyses of miRNA
2.7. Protein Extraction and Western Blot Analyses
2.8. Kinase Activity Profiling
2.9. Growth of Xenograft Tumors in Mice
2.10. Statistical Methods
3. Results
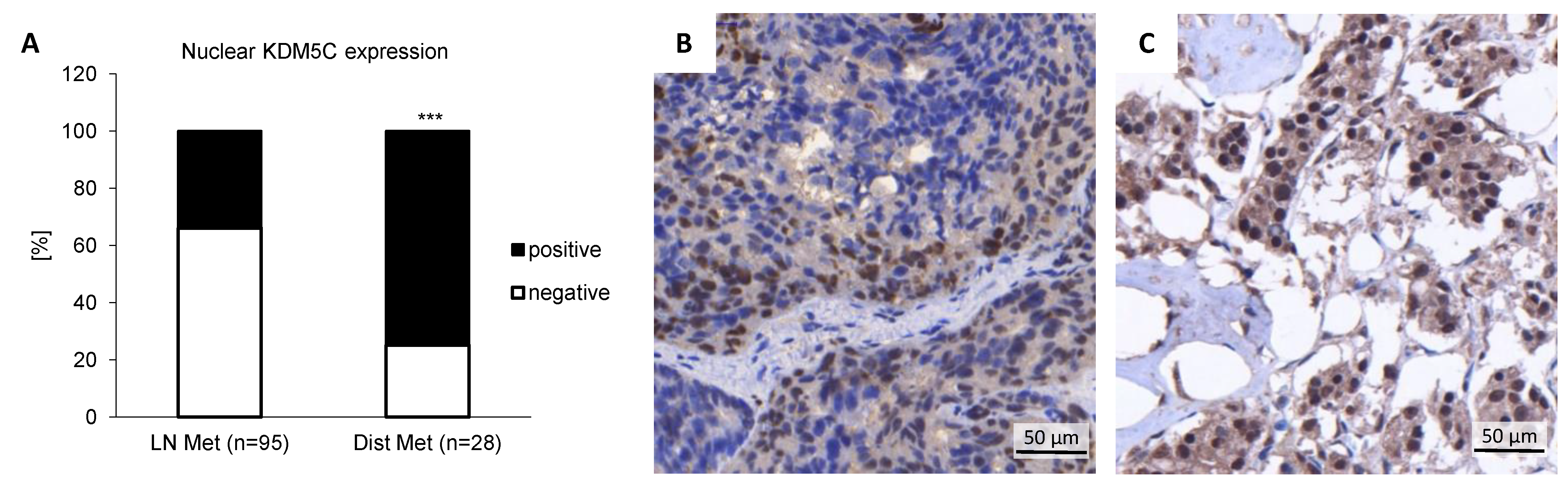
3.1. KDM5C Expression Is Enhanced in Metastatic Prostate Cancer
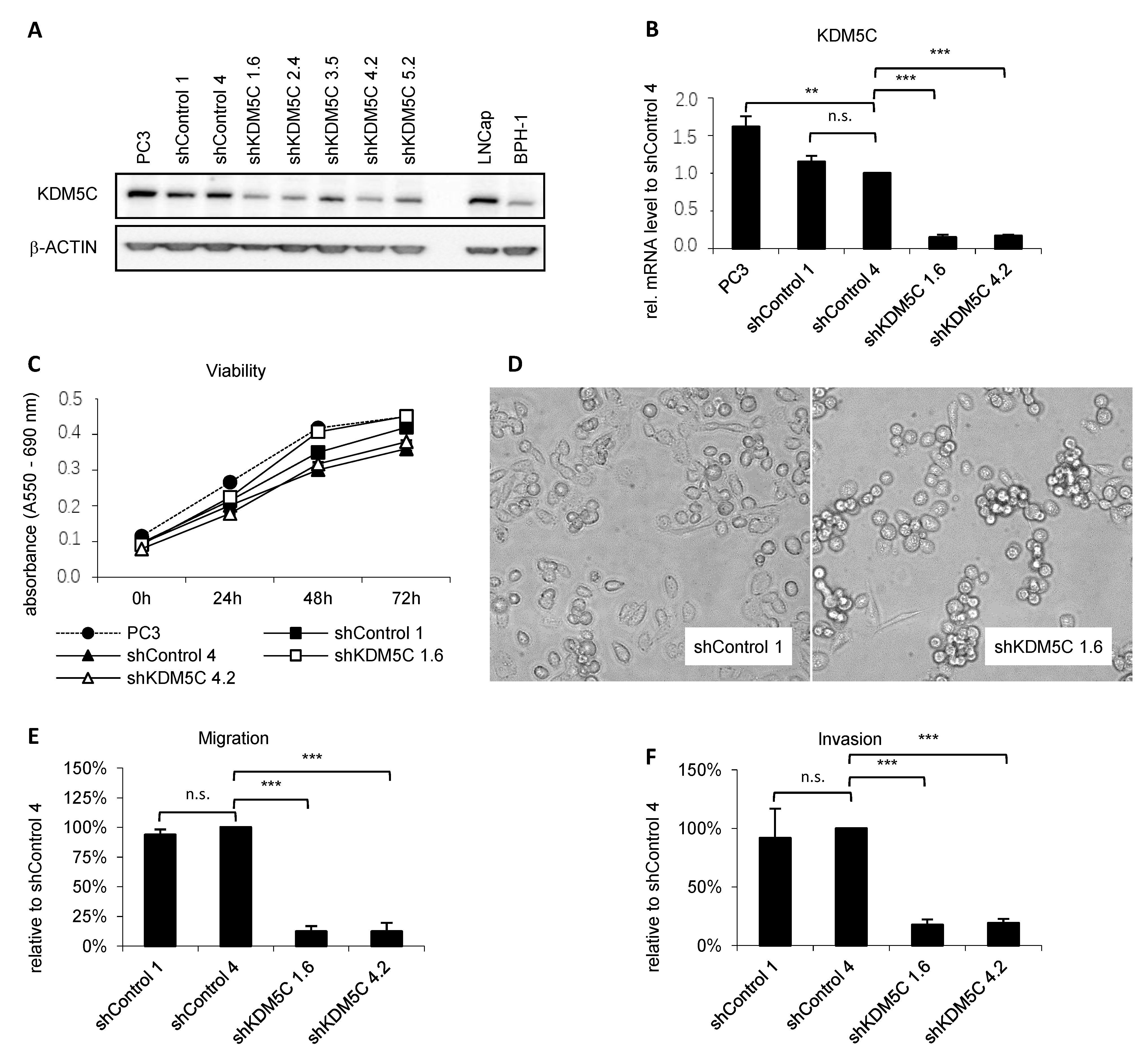
3.2. KDM5C Knockdown Reduces Cell Migration and Invasion
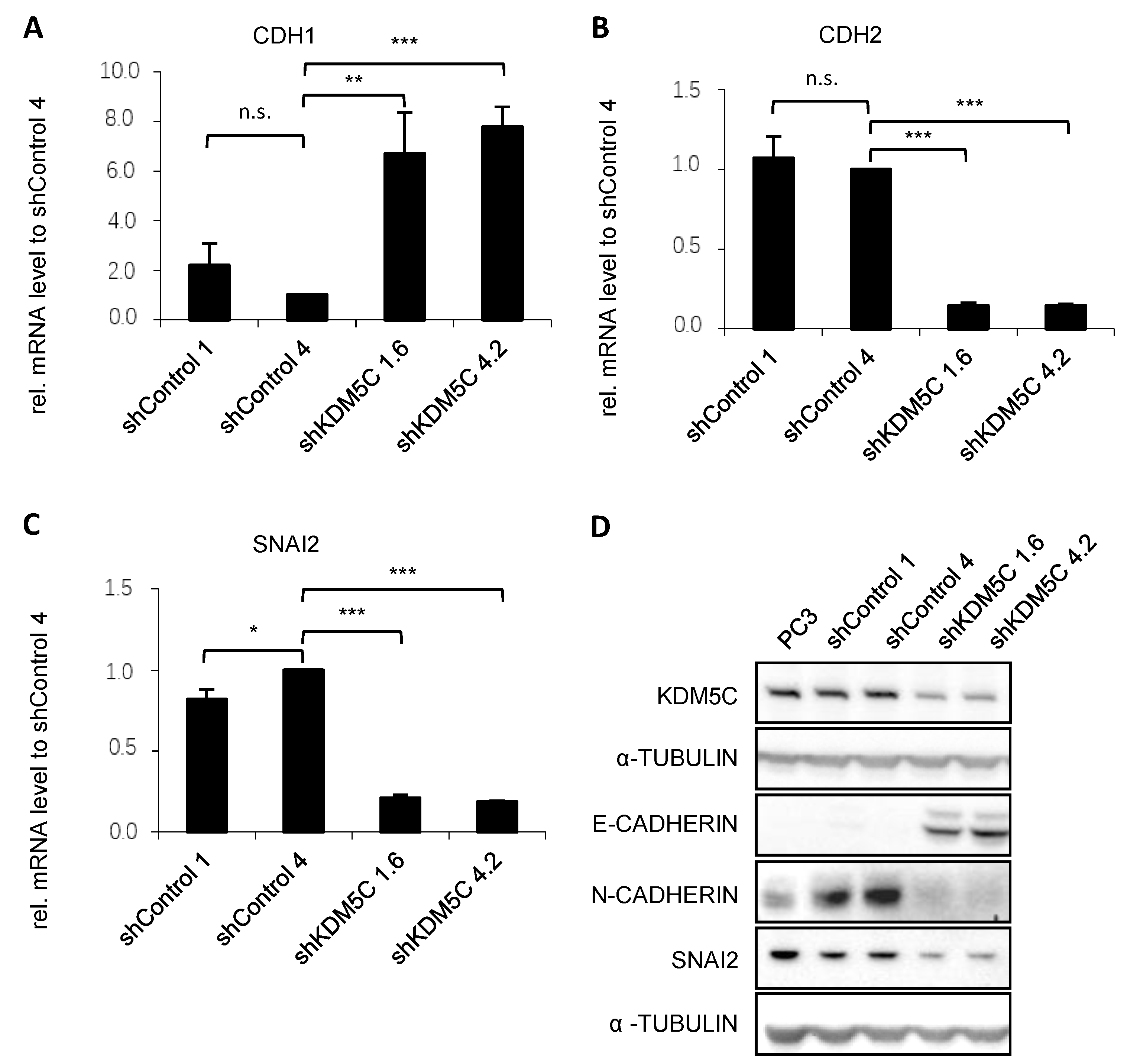
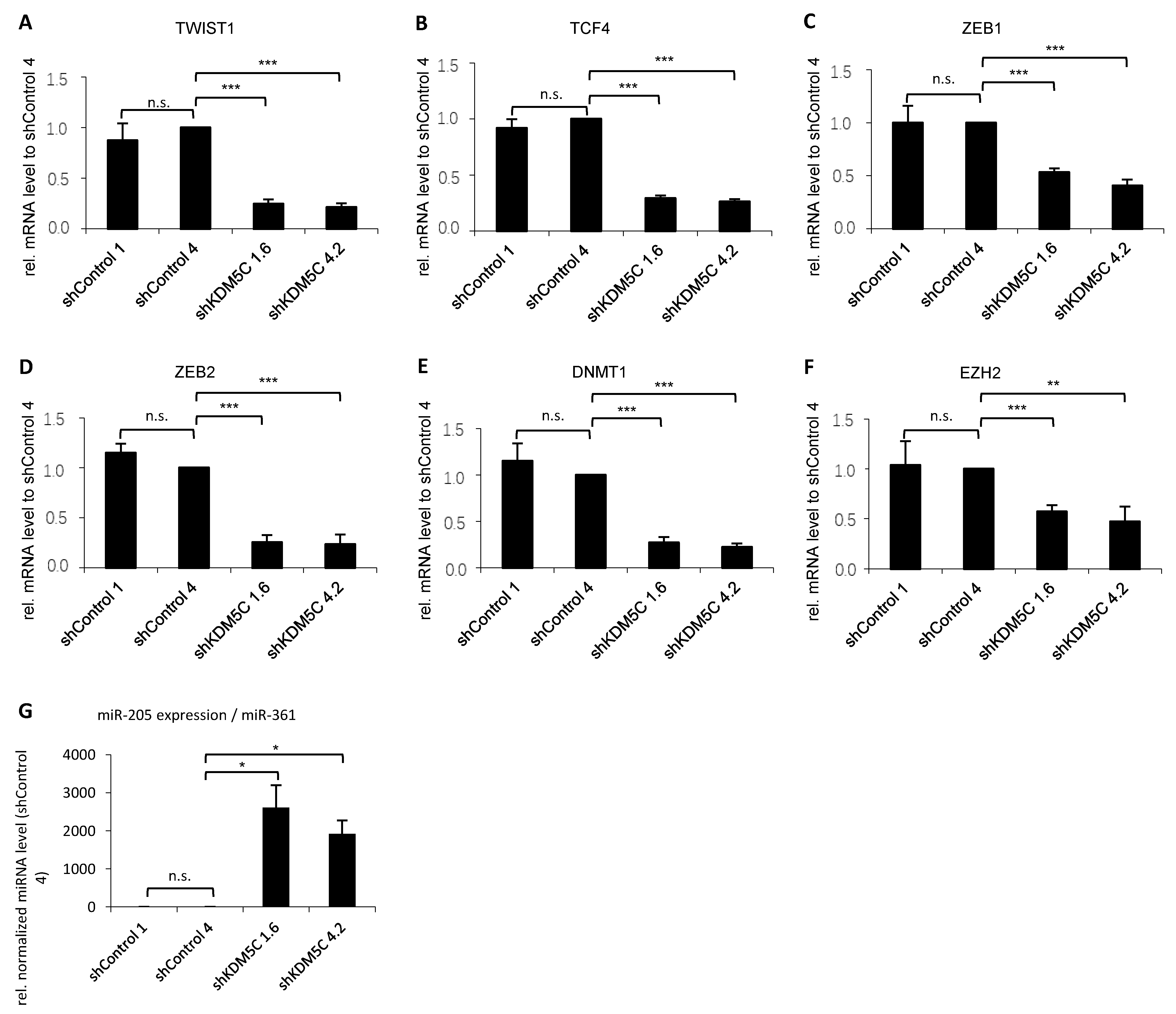
3.3. KDM5C-Mediated Changes in Epithelial-Mesenchymal Transition (EMT)
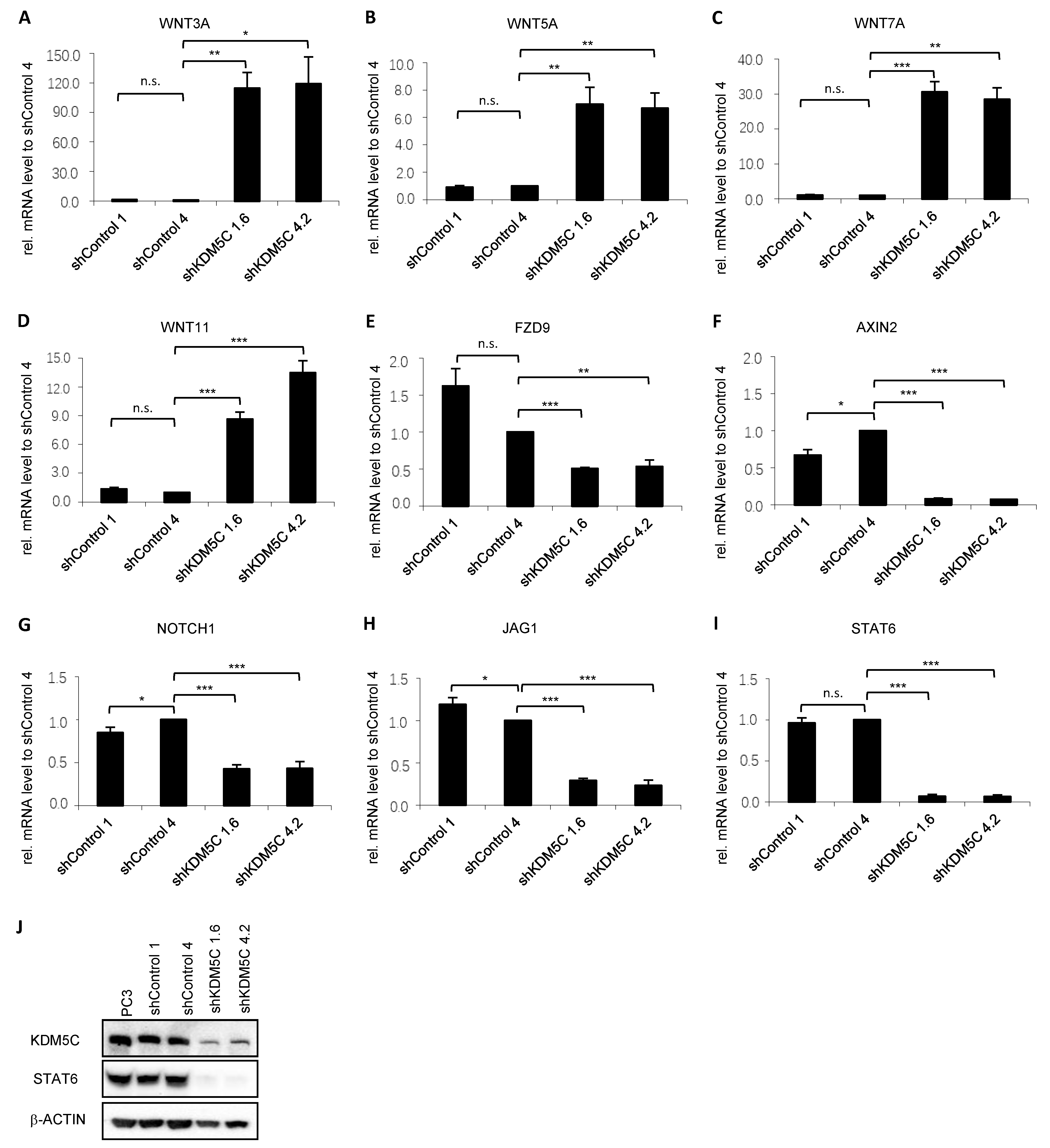
3.4. KDM5C-Mediated Changes in Signaling Pathways
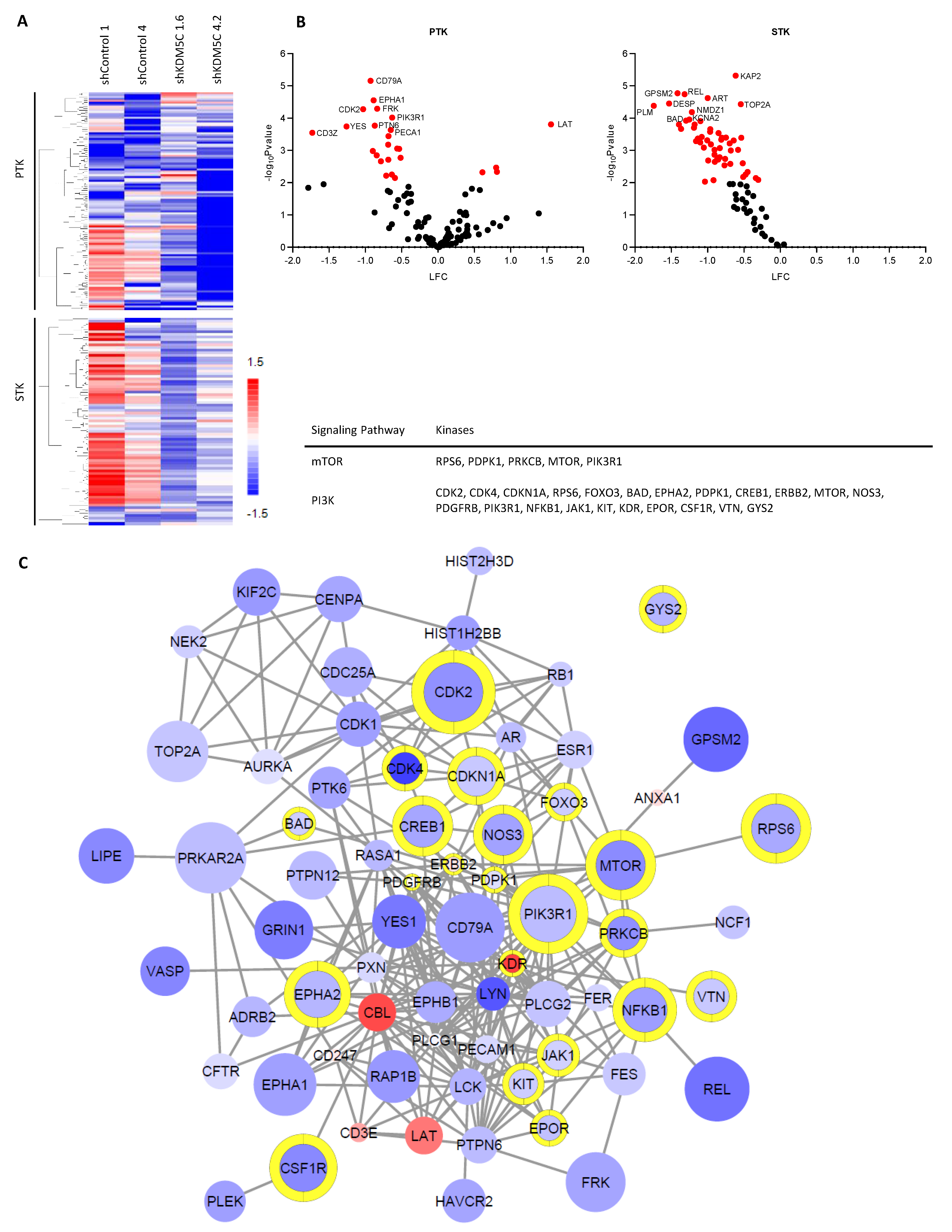
3.5. KDM5C Knockdown Changes Kinase Activity Profile
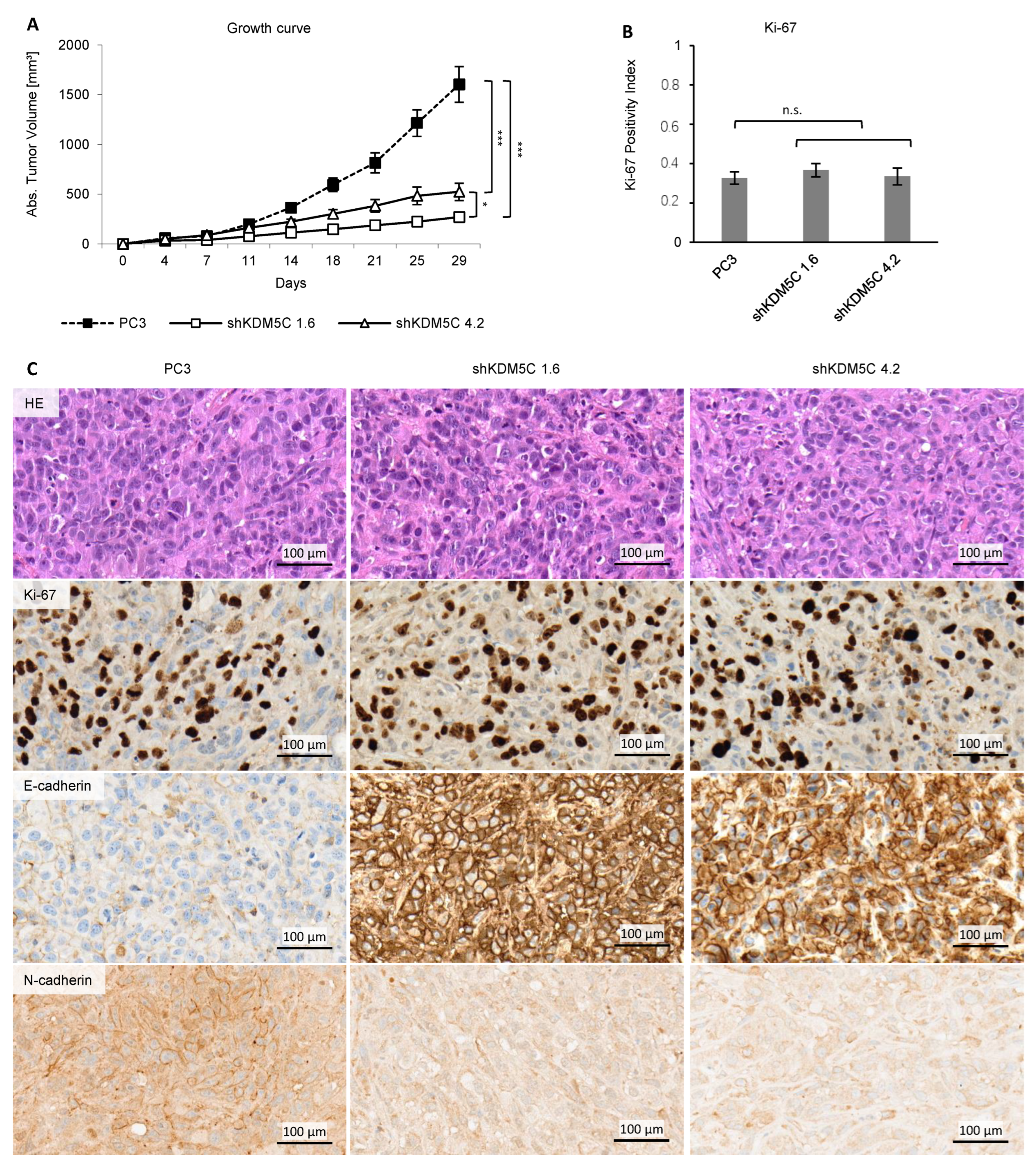
3.6. KDM5C Knockdown Reduces Tumor Growth in Mice
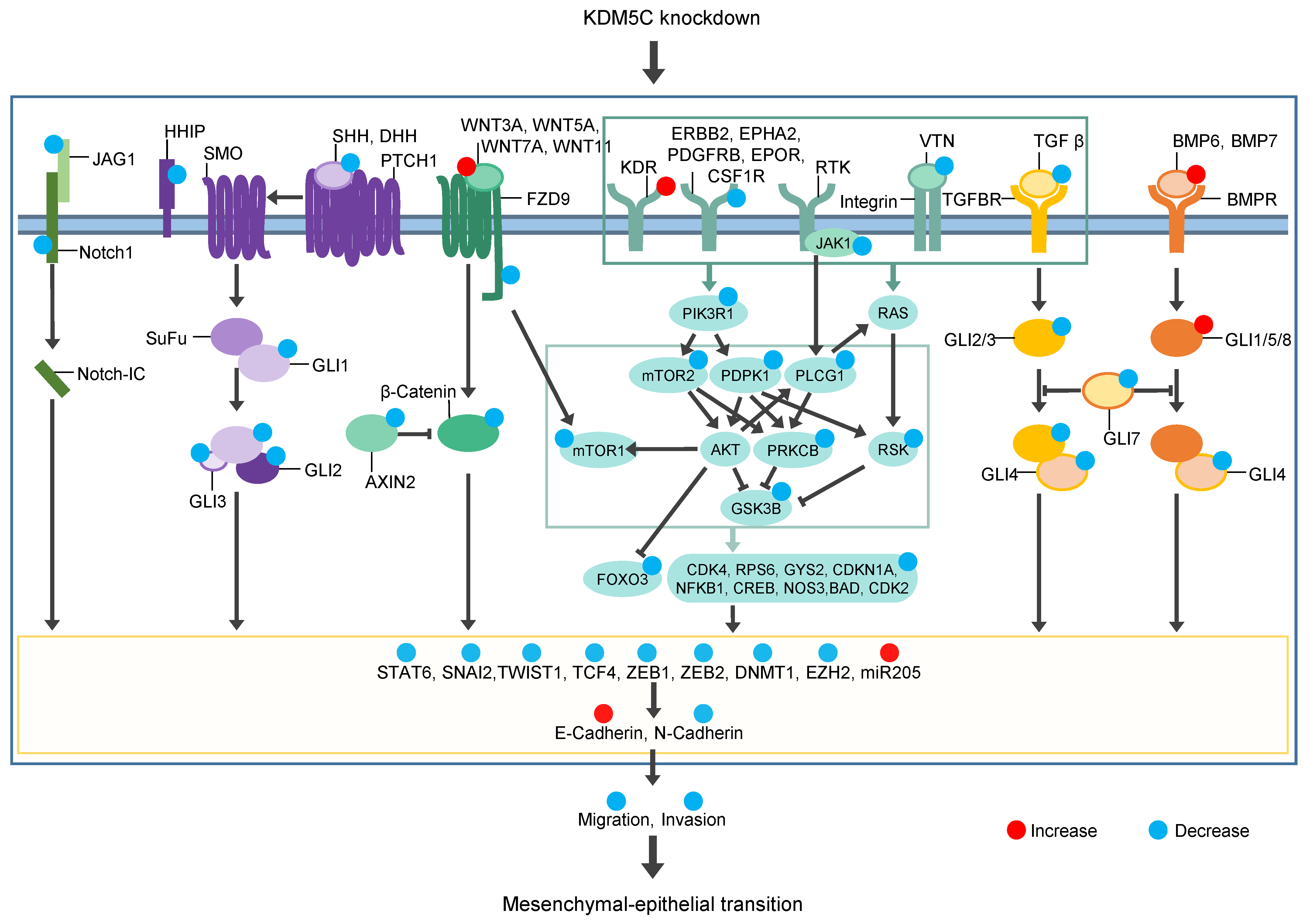
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, D.A.; O′Neil, M.E.; Richards, T.B.; Dowling, N.F.; Weir, H.K. Prostate Cancer Incidence and Survival, by Stage and Race/Ethnicity—United States, 2001–2017. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1473–1480. [Google Scholar] [CrossRef]
- Boutet, A.; De Frutos, C.A.; Maxwell, P.H.; Mayol, M.J.; Romero, J.; Nieto, M.A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. Embo. J. 2006, 25, 5603–5613. [Google Scholar] [CrossRef] [Green Version]
- Berx, G.; Raspe, E.; Christofori, G.; Thiery, J.P.; Sleeman, J.P. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin. Exp. Metastasis 2007, 24, 587–597. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xu, W.; Cheng, P.; Jin, H.; Wang, X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 2017, 8, 8980–8991. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Majores, M.; Rohde, M.; Lim, S.; Schneider, S.; Krappe, E.; Ellinger, J.; Dietel, M.; Stephan, C.; Jung, K.; et al. KDM5C is overexpressed in prostate cancer and is a prognostic marker for prostate-specific antigen-relapse following radical prostatectomy. Am. J. Pathol. 2014, 184, 2430–2437. [Google Scholar] [CrossRef]
- Xu, L.; Wu, W.; Cheng, G.; Qian, M.; Hu, K.; Yin, G.; Wang, S. Enhancement of proliferation and invasion of gastric cancer cell by KDM5C via decrease in p53 expression. Technol. Cancer Res. Treat. 2017, 16, 141–149. [Google Scholar] [CrossRef]
- Ji, X.; Jin, S.; Qu, X.; Li, K.; Wang, H.; He, H.; Guo, F.; Dong, L. Lysine-specific demethylase 5C promotes hepatocellular carcinoma cell invasion through inhibition BMP7 expression. BMC Cancer 2015, 15, 801. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Dhar, S.S.; Chen, T.Y.; Kan, P.Y.; Wei, Y.; Kim, J.H.; Chan, C.H.; Lin, H.K.; Hung, M.C.; Lee, M.G. JARID1D is a suppressor and prognostic marker of prostate cancer invasion and metastasis. Cancer Res. 2016, 76, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Shaikhibrahim, Z.; Offermann, A.; Braun, M.; Menon, R.; Syring, I.; Nowak, M.; Halbach, R.; Vogel, W.; Ruiz, C.; Zellweger, T.; et al. MED12 overexpression is a frequent event in castration-resistant prostate cancer. Endocr-Relat Cancer 2014, 21, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.; Trautmann, M.; Kindler, D.; Huss, S.; Gruenewald, I.; Dirksen, U.; Renner, M.; Mechtersheimer, G.; Pedeutour, F.; Aman, P.; et al. SRC inhibition represents a potential therapeutic strategy in liposarcoma. Int. J. Cancer 2015, 137, 2578–2588. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandellini, P.; Folini, M.; Longoni, N.; Pennati, M.; Binda, M.; Colecchia, M.; Salvioni, R.; Supino, R.; Moretti, R.; Limonta, P.; et al. miR-205 exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res. 2009, 69, 2287–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.L.; Simons, B.W.; Eberhart, C.G.; Berman, D.M. Notch signaling in prostate cancer: A moving target. Prostate 2014, 74, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021, 39, 1150–1162.e9. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol Pharm. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Zaravinos, A. The Regulatory Role of MicroRNAs in EMT and Cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tian, X.J.; Xing, J. Signal transduction pathways of EMT induced by TGF-beta, SHH, and WNT and their crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Schaar, A.; Sukumaran, P.; Dhasarathy, A.; Singh, B.B. TGFbeta-induced epithelial-to-mesenchymal transition in prostate cancer cells is mediated via TRPM7 expression. Mol. Carcinog. 2018, 57, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Park, H.J.; Lee, S.K. The dual role of bone morphogenetic proteins in cancer. Mol. Ther.-Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Yan, J.D.; Yang, S.; Zhang, J.; Zhu, T.H. BMP6 reverses TGF-beta1-induced changes in HK-2 cells: Implications for the treatment of renal fibrosis. Acta Pharmacol. Sin. 2009, 30, 994–1000. [Google Scholar] [CrossRef] [Green Version]
- Buijs, J.T.; Rentsch, C.A.; van der Horst, G.; van Overveld, P.G.; Wetterwald, A.; Schwaninger, R.; Henriquez, N.V.; Ten Dijke, P.; Borovecki, F.; Markwalder, R.; et al. BMP7, a putat.tive regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am. J. Pathol. 2007, 171, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Villarroel, A.; Del Valle-Perez, B.; Fuertes, G.; Curto, J.; Ontiveros, N.; Garcia de Herreros, A.; Dunach, M. Src and Fyn define a new signaling cascade activated by canonical and non-canonical Wnt ligands and required for gene transcription and cell invasion. Cell Mol. Life Sci. 2020, 77, 919–935. [Google Scholar] [CrossRef]
- Sandsmark, E.; Hansen, A.F.; Selnaes, K.M.; Bertilsson, H.; Bofin, A.M.; Wright, A.J.; Viset, T.; Richardsen, E.; Drablos, F.; Bathen, T.F.; et al. A novel non-canonical Wnt signature for prostate cancer aggressiveness. Oncotarget 2017, 8, 9572–9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a signaling in cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jho, E.H.; Zhang, T.; Domon, C.; Joo, C.K.; Freund, J.N.; Costantini, F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell Biol. 2002, 22, 1172–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor′s transmembrane domain. Oncotarget 2017, 8, 91459–91470. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; He, C.S.; Xu, F.; Liu, J.L.; Hu, Z.H.; Zhao, L.P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef]
- Bin Hafeez, B.; Adhami, V.M.; Asim, M.; Siddiqui, I.A.; Bhat, K.M.; Zhong, W.; Saleem, M.; Din, M.; Setaluri, V.; Mukhtar, H. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin. Cancer Res. 2009, 15, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Santagata, S.; Demichelis, F.; Riva, A.; Varambally, S.; Hofer, M.D.; Kutok, J.L.; Kim, R.; Tang, J.; Montie, J.E.; Chinnaiyan, A.M.; et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004, 64, 6854–6857. [Google Scholar] [CrossRef] [Green Version]
- Nanta, R.; Kumar, D.; Meeker, D.; Rodova, M.; Van Veldhuizen, P.J.; Shankar, S.; Srivastava, R.K. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rgamma null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2013, 2, e42. [Google Scholar] [CrossRef]
- Ishii, A.; Shigemura, K.; Kitagawa, K.; Sung, S.Y.; Chen, K.C.; Yi-Te, C.; Liu, M.C.; Fujisawa, M. Anti-tumor Effect of Hedgehog Signaling Inhibitor, Vismodegib, on Castration-resistant Prostate Cancer. Anticancer Res. 2020, 40, 5107–5114. [Google Scholar] [CrossRef]
- Yamamichi, F.; Shigemura, K.; Behnsawy, H.M.; Meligy, F.Y.; Huang, W.C.; Li, X.; Yamanaka, K.; Hanioka, K.; Miyake, H.; Tanaka, K.; et al. Sonic hedgehog and androgen signaling in tumor and stromal compartments drives epithelial-mesenchymal transition in prostate cancer. Scand. J. Urol. 2014, 48, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Nishizaki, Y.; Hui, C.C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [CrossRef]
- Holtz, A.M.; Peterson, K.A.; Nishi, Y.; Morin, S.; Song, J.Y.; Charron, F.; McMahon, A.P.; Allen, B.L. Essential role for ligand-dependent feedback antagonism of vertebrate hedgehog signaling by PTCH1, PTCH2 and HHIP1 during neural patterning. Development 2013, 140, 3423–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, M.K.; Soltani, A.; Soleimani, A.; Kahkhaie, K.R.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cheng, H.; Pan, T.; Liu, Y.; Su, Y.; Ren, C.; Huang, D.; Zha, X.; Liang, C. mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol. Carcinog. 2015, 54, 1086–1095. [Google Scholar] [CrossRef]
- Garcia, G.E.; Nicole, A.; Bhaskaran, S.; Gupta, A.; Kyprianou, N.; Kumar, A.P. Akt-and CREB-mediated prostate cancer cell proliferation inhibition by Nexrutine, a Phellodendron amurense extract. Neoplasia 2006, 8, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Yu, J.; Zhou, Y.; Wang, C.; Jiao, Z.; Qian, Z.; Sun, H.; Chen, B. Identification of CDK2 as a novel target in treatment of prostate cancer. Future Oncol. 2018, 14, 709–718. [Google Scholar] [CrossRef]
- Arai, K.; Eguchi, T.; Rahman, M.M.; Sakamoto, R.; Masuda, N.; Nakatsura, T.; Calderwood, S.K.; Kozaki, K.; Itoh, M. A novel high-throughput 3D screening system for EMT inhibitors: A pilot screening discovered the EMT inhibitory activity of CDK2 inhibitor SU9516. PLoS ONE 2016, 11, e0162394. [Google Scholar] [CrossRef]
- Hussain, S.S.; Huang, S.B.; Bedolla, R.G.; Rivas, P.; Basler, J.W.; Swanson, G.P.; Hui-Ming Huang, T.; Narayanasamy, G.; Papanikolaou, N.; Miyamoto, H.; et al. Suppression of ribosomal protein RPS6KB1 by Nexrutine increases sensitivity of prostate tumors to radiation. Cancer Lett. 2018, 433, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Mazor, M.; Kawano, Y.; Zhu, H.; Waxman, J.; Kypta, R.M. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene 2004, 23, 7882–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, C.R.; Kassis, J.; Whaley, D.L.; Kim, H.G.; Wells, A. PLC gamma contributes to metastasis of in situ-occurring mammary and prostate tumors. Oncogene 2007, 26, 3020–3026. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Lee, Y.C.; Cheng, C.J.; Wu, C.F.; Song, J.H.; Gallick, G.E.; Yu-Lee, L.Y.; Kuang, J.; Lin, S.H. RSK promotes prostate cancer progression in bone through ING3, CKAP2, and PTK6-mediated cell survival. Mol. Cancer Res. 2015, 13, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Maffucci, T.; Raimondi, C.; Abu-Hayyeh, S.; Dominguez, V.; Sala, G.; Zachary, I.; Falasca, M. A phosphoinositide 3-kinase/phospholipase Cgamma1 pathway regulates fibroblast growth factor-induced capillary tube formation. PLoS ONE 2009, 4, e8285. [Google Scholar] [CrossRef]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar] [CrossRef] [Green Version]
- Peltier, J.; O′Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Wang, Z. Akt binds to and phosphorylates phospholipase C-gamma1 in response to epidermal growth factor. Mol. Biol. Cell 2006, 17, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Su, L.; Huang, L.; Song, Z.Y. GSK3beta inhibits epithelial-mesenchymal transition via the Wnt/beta-catenin and PI3K/Akt pathways. Int. J. Ophthalmol. 2018, 11, 1120–1128. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Emadi Baygi, M.; Soheili, Z.S.; Essmann, F.; Deezagi, A.; Engers, R.; Goering, W.; Schulz, W.A. Slug/SNAI2 regulates cell proliferation and invasiveness of metastatic prostate cancer cell lines. Tumour. Biol. 2010, 31, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, K.; O′Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Itsumi, M.; Takeuchi, A.; Imada, K.; Yokomizo, A.; Kuruma, H.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Oda, Y.; et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr. Relat. Cancer 2015, 22, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Tillo, E.; de Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Wang, J.; Yumoto, K.; Jung, Y.; Cackowski, F.C.; Decker, A.M.; Li, Y.; Franceschi, R.T.; Pienta, K.J.; Taichman, R.S. DNMT1 regulates epithelial-mesenchymal transition and cancer stem cells, which promotes prostate cancer metastasis. Neoplasia 2016, 18, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; He, C.; Gao, P.; Wang, S.; Lv, R.; Zhou, H.; Zhou, Q.; Zhang, K.; Sun, J.; Fan, C.; et al. HNF1B-mediated repression of SLUG is suppressed by EZH2 in aggressive prostate cancer. Oncogene 2020, 39, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3117. [Google Scholar] [CrossRef]
- Nishikawa, R.; Goto, Y.; Kurozumi, A.; Matsushita, R.; Enokida, H.; Kojima, S.; Naya, Y.; Nakagawa, M.; Ichikawa, T.; Seki, N. MicroRNA-205 inhibits cancer cell migration and invasion via modulation of centromere protein F regulating pathways in prostate cancer. Int. J. Urol. 2015, 22, 867–877. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemster, A.-L.; Sievers, E.; Pasternack, H.; Lazar-Karsten, P.; Klümper, N.; Sailer, V.; Offermann, A.; Brägelmann, J.; Perner, S.; Kirfel, J. Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT. Cancers 2022, 14, 1894. https://doi.org/10.3390/cancers14081894
Lemster A-L, Sievers E, Pasternack H, Lazar-Karsten P, Klümper N, Sailer V, Offermann A, Brägelmann J, Perner S, Kirfel J. Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT. Cancers. 2022; 14(8):1894. https://doi.org/10.3390/cancers14081894
Chicago/Turabian StyleLemster, Anna-Lena, Elisabeth Sievers, Helen Pasternack, Pamela Lazar-Karsten, Niklas Klümper, Verena Sailer, Anne Offermann, Johannes Brägelmann, Sven Perner, and Jutta Kirfel. 2022. "Histone Demethylase KDM5C Drives Prostate Cancer Progression by Promoting EMT" Cancers 14, no. 8: 1894. https://doi.org/10.3390/cancers14081894