
Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction

2. The Potential of Ferroptosis in (Hepatocellular) Cancer Therapy
2.1. Ferroptosis in Cancer
2.2. The Potential Role of Ferroptosis in Hepatocytes and Liver Pathologies
2.2.1. Hepatocellular Carcinoma
2.2.2. Iron in the Liver
2.2.3. Ferroptosis/Iron Homeostasis in Hepatic Diseases
2.2.4. Iron and PUFAs in HCC
2.2.5. GPX4 and SLC7A11 Expression in HCC
3. The Pharmacological Induction of Ferroptosis in HCC Cells
4. The Translation of Ferroptosis into Clinical HCC Practice
4.1. Ferroptosis Scoring System
4.2. Nanoparticles and Exosomes
4.3. Long Noncoding RNAs/miRNA
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.; Musavi, L.S.; Lee, E.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, M.I.J.; Aichler, M.; Walch, M.A.A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, W.; Bae, K.-H.; Lee, S.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Sassetti, E.; Clausen, M.H.; Laraia, L. Small-Molecule Inhibitors of Reactive Oxygen Species Production. J. Med. Chem. 2021, 64, 5252–5275. [Google Scholar] [CrossRef]
- Clemente, L.P.; Rabenau, M.; Tang, S.; Stanka, J.; Cors, E.; Stroh, J.; Culmsee, C.; Von Karstedt, S. Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration. Cells 2020, 9, 2259. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2018, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Mumbauer, S.; Pascual, J.; Kolotuev, I.; Hamaratoglu, F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019, 15, e1008396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.-D.; Pang, P.; Zhou, X.-T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.-Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Geng, N.; Shi, B.-J.; Li, S.-L.; Zhong, Z.-Y.; Li, Y.-C.; Xua, W.-L.; Zhou, H.; Cai, J.-H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e574. [Google Scholar] [CrossRef]
- Protchenko, O.; Baratz, E.; Jadhav, S.; Li, F.; Shakoury-Elizeh, M.; Gavrilova, O.; Ghosh, M.C.; Cox, J.E.; Maschek, J.A.; Tyurin, V.A.; et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology 2021, 73, 1176–1193. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021, 28, 2843–2856. [Google Scholar] [CrossRef]
- Reinhardt-Rutland, A.H. Interaural transfer of aftereffect of changing sound level in a tone. J. Gen. Psychol. 1988, 115, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhu, J.-Y.; Zang, X.; Zhai, Y.-Z. The Emerging Role of Ferroptosis in Liver Diseases. Front. Cell Dev. Biol. 2021, 9, 801365. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, M.A.; Behnke, C.A.; Eastman, A. Activation of programmed cell death (apoptosis) by cisplatin, other anticancer drugs, toxins and hyperthermia. Biochem. Pharmacol. 1990, 40, 2353–2362. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, K.; Yanatori, I.; Hara, Y.; Nishina, S. Iron and liver cancer: An inseparable connection. FEBS J. 2021. [Google Scholar] [CrossRef]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Front. Oncol. 2021, 11, 778492. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2016, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Liao, H.; Shi, J.; Wen, K.; Lin, J.; Liu, Q.; Shi, B.; Yan, Y.; Xiao, Z. Molecular Targets of Ferroptosis in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.-Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.-H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, G.M. Sorafenib: A Review in Hepatocellular Carcinoma. Target. Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar®), a Dual-Action Inhibitor That Targets RAF/MEK/ERK Pathway in Tumor Cells and Tyrosine Kinases VEGFR/PDGFR in Tumor Vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’H, K. Ferroptosis in Liver Diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 73, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Correnti, M.; Gammella, E.; Raggi, C.; Invernizzi, P.; Cairo, G. Iron Metabolism in Liver Cancer Stem Cells. Front. Oncol. 2019, 9, 149. [Google Scholar] [CrossRef]
- Lim, K.; Han, C.; Dai, Y.; Shen, M.; Wu, T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking β-catenin and cyclooxygenase-2. Mol. Cancer Ther. 2009, 8, 3046–3055. [Google Scholar] [CrossRef] [Green Version]
- Weylandt, K.H.; Krause, L.F.; Gomolka, B.; Chiu, C.-Y.; Bilal, S.; Nadolny, A.; Waechter, S.F.; Fischer, A.; Rothe, M.; Kang, J.X. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-. Carcinogenesis 2011, 32, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef]
- Guerriero, E.; Capone, F.; Accardo, M.; Sorice, A.; Costantini, M.; Colonna, G.; Castello, G. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur. J. Histochem. 2015, 59, 2540. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc- cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2015, 23, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar]
- Li, Y.; Xia, J.; Shao, F.; Zhou, Y.; Yu, J.; Wu, H.; Du, J.; Ren, X. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem. Biophys. Res. Commun. 2020, 534, 877–884. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; Francois, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.-C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Werth, E.; Rajbhandari, P.; Stockwell, B.R.; Brown, L.M. Time Course of Changes in Sorafenib-Treated Hepatocellular Carcinoma Cells Suggests Involvement of Phospho-Regulated Signaling in Ferroptosis Induction. Proteomics 2020, 20, e2000006. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhou, C.; Zhao, Y.; Zhang, X.; Chen, W.; Zhou, Q.; Hu, B.; Gao, D.; Raatz, L.; Wang, Z.; et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021, 41, 101942. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-J.; Dai, H.-Q.; Huang, X.-W.; Feng, J.; Deng, J.-H.; Wang, Z.-X.; Yang, X.-M.; Liu, Y.-J.; Wu, Y.; Chen, P.-H.; et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol. Sin. 2020, 42, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, J.; Petri, K.; Fulda, S.; Liese, J. Redox Modulation and Induction of Ferroptosis as a New Therapeutic Strategy in Hepatocellular Carcinoma. Transl. Oncol. 2020, 13, 100785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef]
- Lin, P.-L.; Tang, H.-H.; Wu, S.-Y.; Shaw, N.-S.; Su, A.C.-L. Saponin Formosanin C-induced Ferritinophagy and Ferroptosis in Human Hepatocellular Carcinoma Cellsa. Antioxidants 2020, 9, 682. [Google Scholar] [CrossRef]
- Chang, W.-T.; Bow, Y.-D.; Fu, P.-J.; Li, C.-Y.; Wu, C.-Y.; Chang, Y.-H.; Teng, Y.-N.; Li, R.-N.; Lu, M.-C.; Liu, Y.-C.; et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef]
- Jin, M.; Shi, C.; Li, T.; Wu, Y.; Hu, C.; Huang, G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed. Pharmacother. 2020, 129, 110282. [Google Scholar] [CrossRef]
- Huang, C.; Santofimia-Castaño, P.; Liu, X.; Xia, Y.; Peng, L.; Gotorbe, C.; Neira, J.L.; Tang, D.; Pouyssegur, J.; Iovanna, J. NUPR1 inhibitor ZZW-115 induces ferroptosis in a mitochondria-dependent manner. Cell Death Discov. 2021, 7, 269. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Lei, P.; Zhou, H.; Liang, R.; Zhu, R.; Wang, W.; Zhou, L.; Sun, Y. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J. Cell. Mol. Med. 2019, 23, 7349–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Heiden, M.G.V.; McCormick, F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Asadi, M.R.; Moslehian, M.S.; Sabaie, H.; Poornabi, M.; Ghasemi, E.; Hassani, M.; Hussen, B.M.; Taheri, M.; Rezazadeh, M. Stress Granules in the Anti-Cancer Medications Mechanism of Action: A Systematic Scoping Review. Front. Oncol. 2021, 11, 797549. [Google Scholar] [CrossRef]
- Mao, C.; Wang, X.; Liu, Y.; Wang, M.; Yan, B.; Jiang, Y.; Shi, Y.; Shen, Y.; Liu, X.; Liai, W.; et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018, 78, 3484–3496. [Google Scholar] [CrossRef] [Green Version]
- Dolicka, D.; Foti, M.; Sobolewski, C. The Emerging Role of Stress Granules in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 9428. [Google Scholar] [CrossRef]
- Zhu, H.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.-H.; Ao, X.; Haifeng, Z.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol. Cancer Res. 2015, 13, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, J.; Deng, H.; Wang, Y.; Lei, C.; Wang, Q.; Xiang, J.; Liang, L.; Xia, J.; Pan, X.; et al. GSTZ 1-1 Deficiency Activates NRF 2/ IGF 1R Axis in HCC via Accumulation of Oncometabolite Succinylacetone. EMBO J. 2019, 38, e101964. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Yang, Y.; Lei, C.; Yang, F.; Liang, L.; Chen, C.; Xia, J.; Wang, K.; Tang, N. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 438. [Google Scholar] [CrossRef]
- Asperti, M.; Bellini, S.; Grillo, E.; Gryzik, M.; Cantamessa, L.; Ronca, R.; Maccarinelli, F.; Salvi, A.; De Petro, G.; Arosio, P.; et al. H-ferritin suppression and pronounced mitochondrial respiration make Hepatocellular Carcinoma cells sensitive to RSL3-induced ferroptosis. Free Radic. Biol. Med. 2021, 169, 294–303. [Google Scholar] [CrossRef]
- Liu, J.; Song, X.; Kuang, F.; Zhang, Q.; Xie, Y.; Kang, R.; Kroemer, G.; Tang, D. NUPR1 is a critical repressor of ferroptosis. Nat. Commun. 2021, 12, 647. [Google Scholar] [CrossRef]
- Bai, T.; Wang, S.; Zhao, Y.; Zhu, R.; Wang, W.; Sun, Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 491, 919–925. [Google Scholar] [CrossRef]
- Tang, H.; Chen, D.; Lia, C.; Zhengc, C.; Wud, X.; Zhanga, Y.; Songc, Q.; Feic, W. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int. J. Pharm. 2019, 572, 118782. [Google Scholar] [CrossRef]
- Ou, W.; Mulik, R.S.; Anwar, A.; McDonald, J.G.; He, X.; Corbin, I.R. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic. Biol. Med. 2017, 112, 597–607. [Google Scholar] [CrossRef]
- Du, J.; Wan, Z.; Wang, C.; Lu, F.; Wei, M.; Wang, D.; Hao, Q. Designer exosomes for targeted and efficient ferroptosis induction in cancer via chemo-photodynamic therapy. Theranostics 2021, 11, 8185–8196. [Google Scholar] [CrossRef]
- Gao, L.; Xue, J.; Liu, X.; Cao, L.; Wang, R.; Lei, L. A scoring model based on ferroptosis genes for prognosis and immunotherapy response prediction and tumor microenvironment evaluation in liver hepatocellular carcinoma. Aging 2021, 13, 24866–24881. [Google Scholar] [CrossRef]
- Deng, T.; Hu, B.; Jin, C.; Tong, Y.; Zhao, J.; Shi, Z.; Zhang, T.; Deng, L.; Sun, Z.; Chen, G.; et al. A novel ferroptosis phenotype-related clinical-molecular prognostic signature for hepatocellular carcinoma. J. Cell. Mol. Med. 2021, 25, 6618–6633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, L.; Liu, L.; Lu, T.; Jiao, D.; Sun, Y.; Han, X. The Identification and Validation of Two Heterogenous Subtypes and a Risk Signature Based on Ferroptosis in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 619242. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Zhang, J.; Tan, J.; Li, J.; Song, Z. Development and Validation of a Combined Ferroptosis and Immune Prognostic Classifier for Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 596679. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-A.; Tian, H.; Yao, D.-M.; Zhang, Y.; Feng, Z.-J.; Yang, C.-J. Identification of a Ferroptosis-Related Signature Model Including mRNAs and lncRNAs for Predicting Prognosis and Immune Activity in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 738477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-J.; Huang, Y.-P.; Li, X.-X.; Liu, Z.-T.; Liu, K.; Deng, X.-F.; Xiong, L.; Zou, H.; Wen, Y. A Novel Ferroptosis-Related 4-Gene Prognostic Signature for Cholangiocarcinoma and Photodynamic Therapy. Front. Oncol. 2021, 11, 747445. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Mayr, C.; Kiesslich, T.; Stintzing, S.; Neureiter, D. Immunmodulatory Treatment Strategies of Hepatocellular Carcinoma: From Checkpoint Inhibitors Now to an Integrated Approach in the Future. Cancers 2021, 13, 1558. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lu, P.-Z.; Zhu, G.-Z.; Hooi, S.C.; Wu, Y.; Huang, X.-W.; Dai, H.-Q.; Chen, P.-H.; Li, Z.-J.; Su, W.-J.; et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol. Sin. 2020, 42, 160–170. [Google Scholar] [CrossRef]
- Neureiter, D.; Stintzing, S.; Kiesslich, T.; Ocker, M. Hepatocellular carcinoma: Therapeutic advances in signaling, epigenetic and immune targets. World J. Gastroenterol. 2019, 25, 3136–3150. [Google Scholar] [CrossRef]
- Zhou, Q.-M.; Lu, Y.-F.; Zhou, J.-P.; Yang, X.-Y.; Wang, X.-J.; Yu, J.-N.; Du, Y.-Z.; Yu, R.-S. Self-amplification of oxidative stress with tumour microenvironment-activatable iron-doped nanoplatform for targeting hepatocellular carcinoma synergistic cascade therapy and diagnosis. J. Nanobiotechnol. 2021, 19, 361. [Google Scholar] [CrossRef]
- Xu, Q.; Zhan, G.; Zhang, Z.; Yong, T.; Yang, X.; Gan, L. Manganese porphyrin-based metal-organic framework for synergistic sonodynamic therapy and ferroptosis in hypoxic tumors. Theranostics 2021, 11, 1937–1952. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhao, S.; Nice, E.C.; Huang, C.; He, W.; Zou, B.; Lin, J. A cascaded copper-based nanocatalyst by modulating glutathione and cyclooxygenase-2 for hepatocellular carcinoma therapy. J. Colloid Interface Sci. 2021, 607, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Li, T.; Xie, X.; Xia, J. Computational Identification of Immune- and Ferroptosis-Related LncRNA Signature for Prognosis of Hepatocellular Carcinoma. Front. Mol. Biosci. 2021, 8, 759173. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Liang, R.; Zhu, R.; Wang, W.; Zhou, L.; Sun, Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J. Cell. Physiol. 2020, 235, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ge, X.; Zhang, Z.; Ye, Y.; Zhou, Z.; Li, M.; Yan, H.; Wu, L.; Bai, Q.; Li, J.; et al. Identification of a Ferroptosis-Related Long Noncoding RNA Prognostic Signature and Its Predictive Ability to Immunotherapy in Hepatocellular Carcinoma. Front. Genet. 2021, 12, 682082. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, T.; Qi, J.; Qin, C.; Zhu, Q. Four Autophagy-Related lncRNAs Predict the Prognosis of HCC through Coexpression and ceRNA Mechanism. BioMed Res. Int. 2020, 2020, 3801748. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Lyu, N.; Zeng, Y.; Kong, Y.; Chen, Q.; Deng, H.; Ou, S.; Bai, Y.; Tang, H.; Wang, X.; Zhao, M. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann. Transl. Med. 2021, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Q.; Wang, X.; Xu, Z.; Wei, X.; Li, J. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein ALKBH5. Cell Death Discov. 2020, 6, 3801748. [Google Scholar] [CrossRef]
- Li, B.; Yang, L.; Peng, X.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Jin, H.; Wu, B.; Huang, M.; et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed. Pharmacother. 2020, 130, 110710. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Zhang, H.; Qin, Q.; Hou, Y.; Zhang, X.; Chen, D.; Zhang, H.; Chen, Y. Ferroptosis as a new therapeutic opportunity for nonviral liver disease. Eur. J. Pharmacol. 2021, 908, 174319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xing, X.; Liu, H.; Feng, J.; Tian, M.; Chang, S.; Liu, P.; Zhang, H. Ionizing radiation induces ferroptosis in granulocyte-macrophage hematopoietic progenitor cells of murine bone marrow. Int. J. Radiat. Biol. 2020, 96, 584–595. [Google Scholar] [CrossRef] [PubMed]



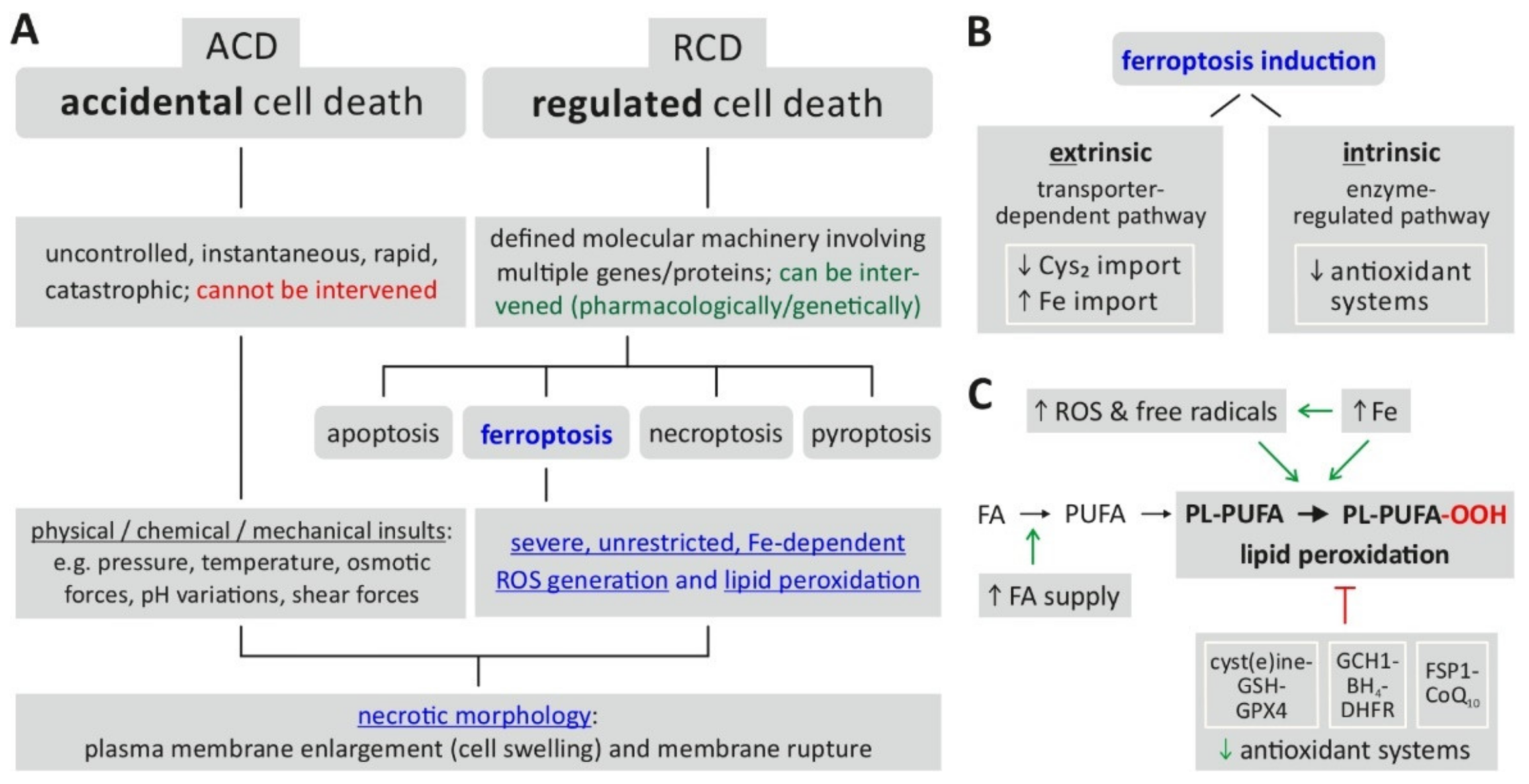{kind=link}
{kind=link}
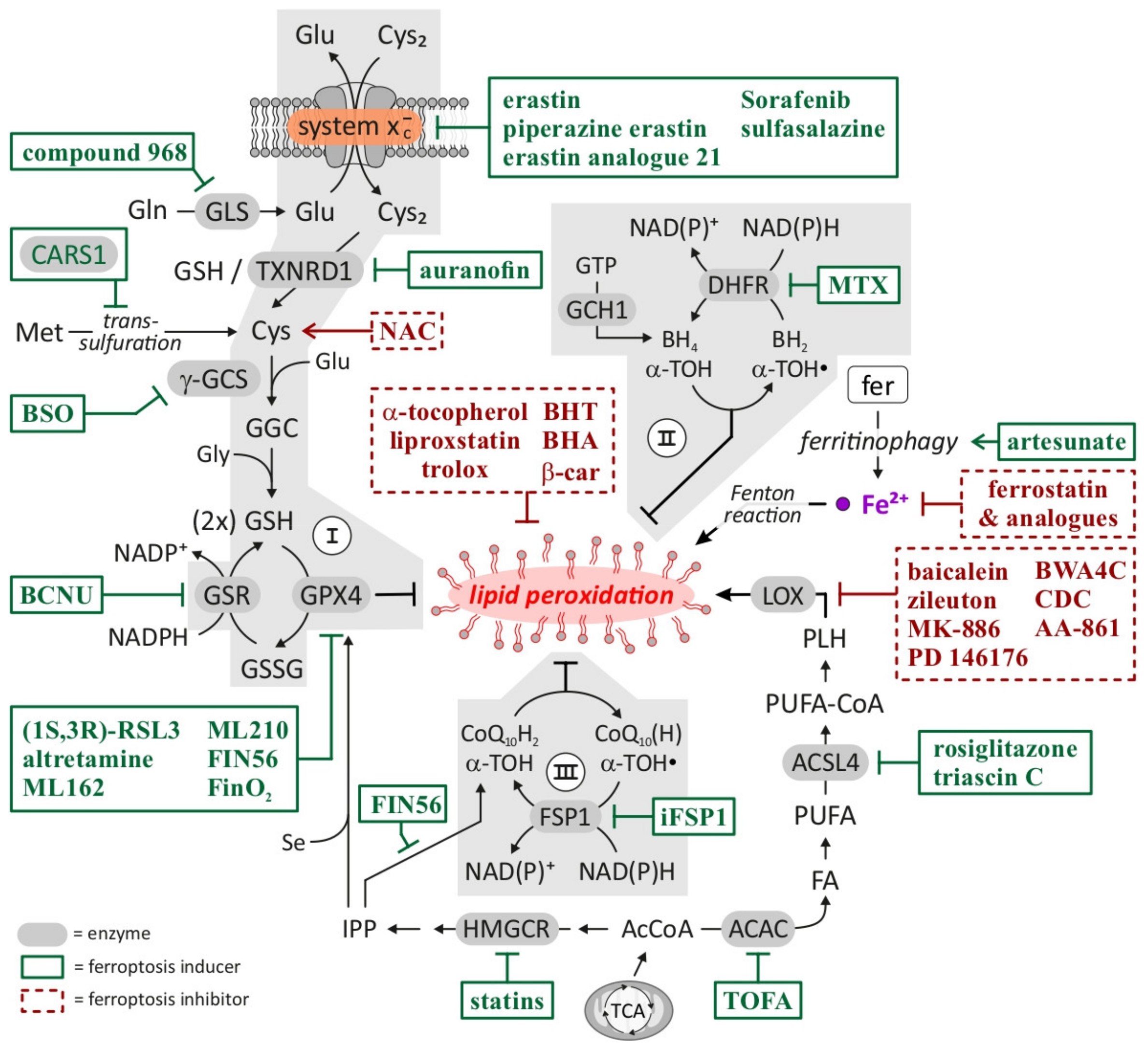{kind=link}
| Substance | Drug Name or Synonym a | Mode of Action | In-Vitro/In-Vivo/In-Situ | References |
|---|---|---|---|---|
| Sorafenib b | Nexavar® | Inhibition of system xc- | ✓/-/- | [78,79,80,81,82,83] |
| Quiescin Sulfhydryl Oxidase 1 | QSOX1 | Inhibition of NRF2 | ✓/✓/- | [84] |
| Artesunate | Arsumax | Ferritin degradation (in combination with sorafenib) | ✓/✓/- | [85] |
| Eradicator of RAS and ST-expressing cells | Erastin | Inhibition of system xc- | ✓/-/- | [86] |
| Ras-selective lethal small molecule 3 | RSL-3 | Inhibition of GPX4 | ✓/✓/- | [87] |
| Saponin Formosanin C | NSC 306864 | Induction of ferritinophagy | ✓/-/- | [88] |
| Heteronemin | CHEMBL514498 | Reduction of GPX4 expression | ✓/✓/- | [89] |
| Solasonine | Tomatine solaradixine | Suppression of GPX4 | ✓/✓/- | [90] |
| ZZW-115 | HY-111838 | Inhibition of NUPR1 | ✓/✓/- | [91] |
| Haloperidol | Haldol® | Inhibition of S1R | ✓/✓/- | [92] |
| Year | Database(s) (Dataset) | Basic Cluster Description | Predictive and/or Prognostic Aspects of the Ferroptosis Cluster | Ref |
|---|---|---|---|---|
| 2021 | TCGA: LIHC GEO: GSE76427 | Ferrcluster A: “Olfactory transduction” and “cardiac music contraction”. Ferrcluster B: “mTOR signaling pathway” and “neurotrophin signaling pathway”. Ferrcluster C: “adipokine signaling pathway”, “tyrosine metabolism” and “PPAR signaling pathway” | Ferrcluster B: Overall survival ↓ High ferrscore group: Survival ↓, Programmed cell death 1 (PD-1) mRNA expression ↑, efficacy of PD-1 or PD-1 plus CTLA4 (cytotoxic T-lymphocyte associated protein 4) inhibitors ↓. | [112] |
| 2021 | TCGA ICGC | Ferroptosis-H and Ferroptosis-L: According to ferroptosis gene expression and methylation | Ferroptosis-H: Overall and disease-specific survival ↓ | [113] |
| 2021 | GEO TCGA ICGC | C1: Metabolism low, immunity high subtype. C2: Metabolism high, immunity low subtype. | C1: Prognosis ↓ C1: Patients with clinical characteristics such as younger, female, advanced stage, higher grade, vascular invasion. | [114] |
| 2020 | GEO: GSE14520/GPL3921 TCGA | Low and high group: Comprehensive index of ferroptosis and immune status (CIFI). | High CIFI: Prognosis ↓ | [115] |
| 2021 | TCGA | Low-risk and high-risk groups: 2 ferroptosis-related mRNAs and ferroptosis-related lncRNAs | Higher risk group: Prognosis ↓ Higher risk group: Differences of tumor microenvironment, immune cell infiltration as well as tumor-related pathways | [116] |
| 2021 a | TCGA-CHOL GEO: GSE107943 EMBL-EBI: E-MTAB-6389 | Low and high group: Ferroptosis-related weighted coexpression gene network and model construction. | Higher risk group: Prognosis ↓ | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekric, D.; Ocker, M.; Mayr, C.; Stintzing, S.; Ritter, M.; Kiesslich, T.; Neureiter, D. Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation. Cancers 2022, 14, 1826. https://doi.org/10.3390/cancers14071826
Bekric D, Ocker M, Mayr C, Stintzing S, Ritter M, Kiesslich T, Neureiter D. Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation. Cancers. 2022; 14(7):1826. https://doi.org/10.3390/cancers14071826
Chicago/Turabian StyleBekric, Dino, Matthias Ocker, Christian Mayr, Sebastian Stintzing, Markus Ritter, Tobias Kiesslich, and Daniel Neureiter. 2022. "Ferroptosis in Hepatocellular Carcinoma: Mechanisms, Drug Targets and Approaches to Clinical Translation" Cancers 14, no. 7: 1826. https://doi.org/10.3390/cancers14071826