
Enhanced T-Cell Priming and Improved Anti-Tumor Immunity through Lymphatic Delivery of Checkpoint Blockade Immunotherapy

 , ,
, ,  and
and 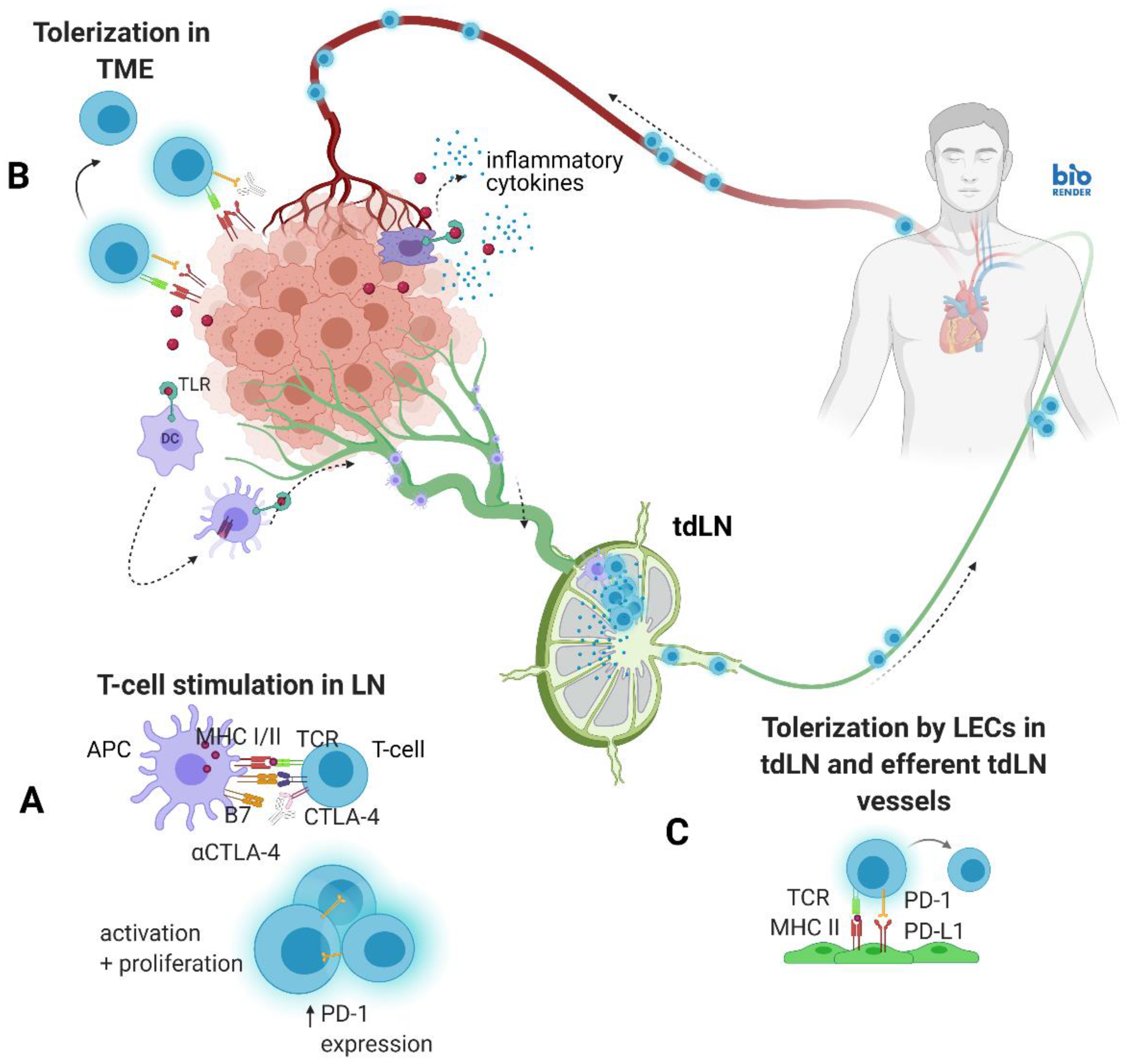{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents and Therapies
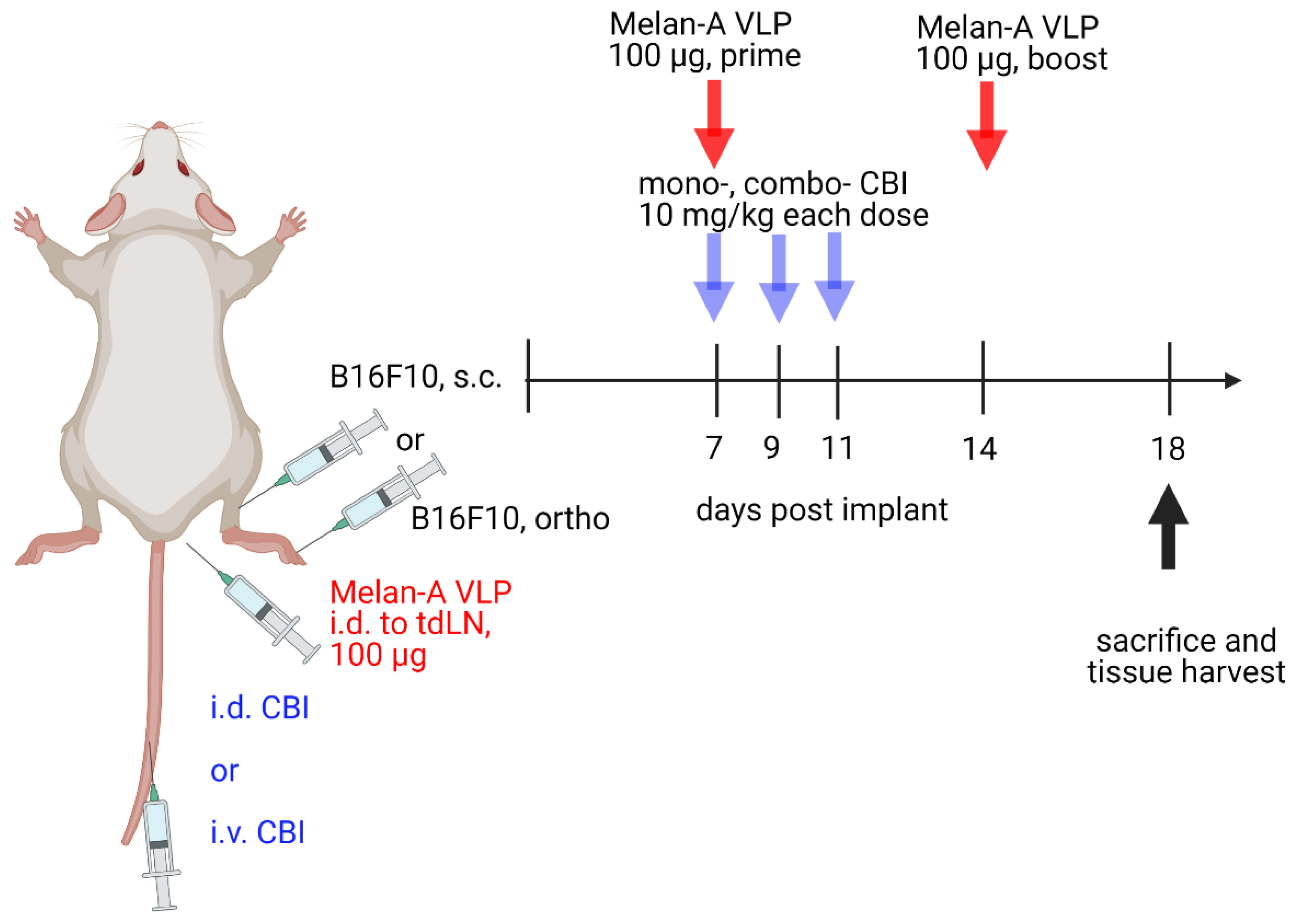
2.3. B16F10 Tumor Models and Experimental Treatment Groups
2.4. RNA Analysis
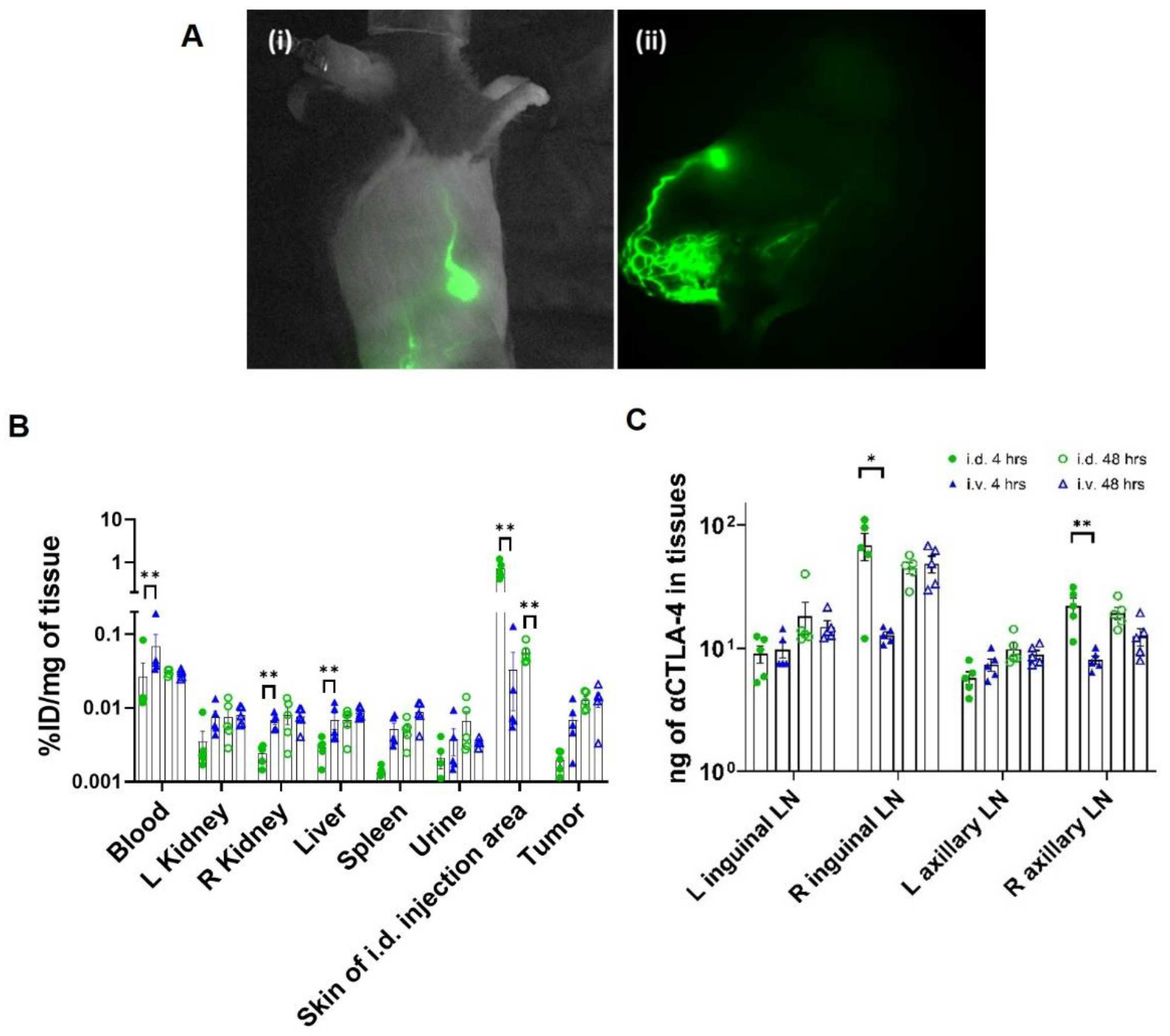
2.5. Confirmation of Lymphatic Delivery via Near-Infrared Fluorescence Imaging (NIRF-LI) and Biodistribution
2.6. Flow Cytometry
2.7. Melan-A Specific Functionality
2.8. Multiplex Immunofluorescence
2.9. Statistics
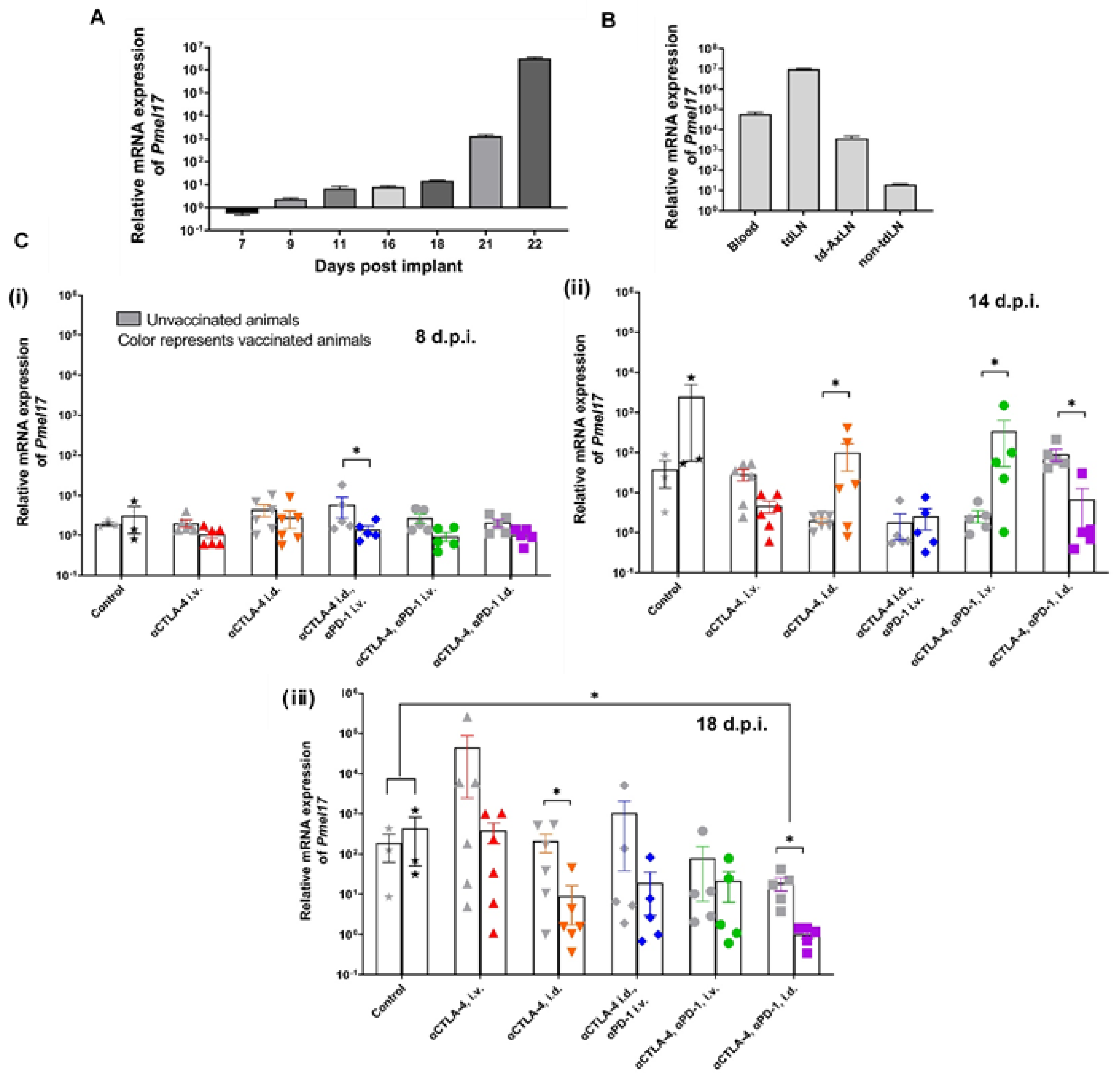
3. Results
3.1. CBI Delivery to the Right Lymphatic Watershed following i.d. Injection at Right Lateral Side of Tail
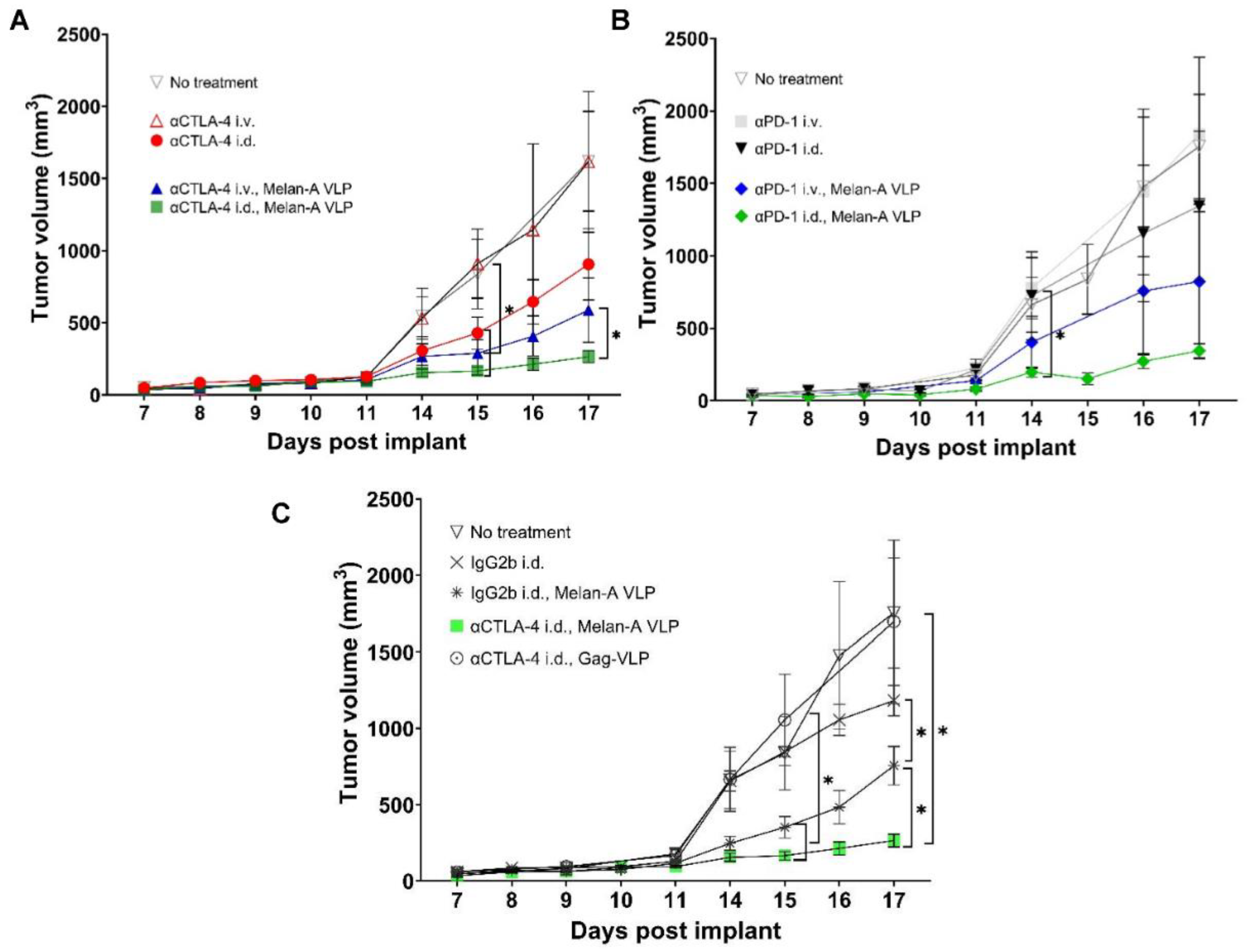
3.2. Tumor Growth Rates Significantly Reduced with i.d. but Not i.v. Delivery of CBI Monotherapy in Vaccinated Animals
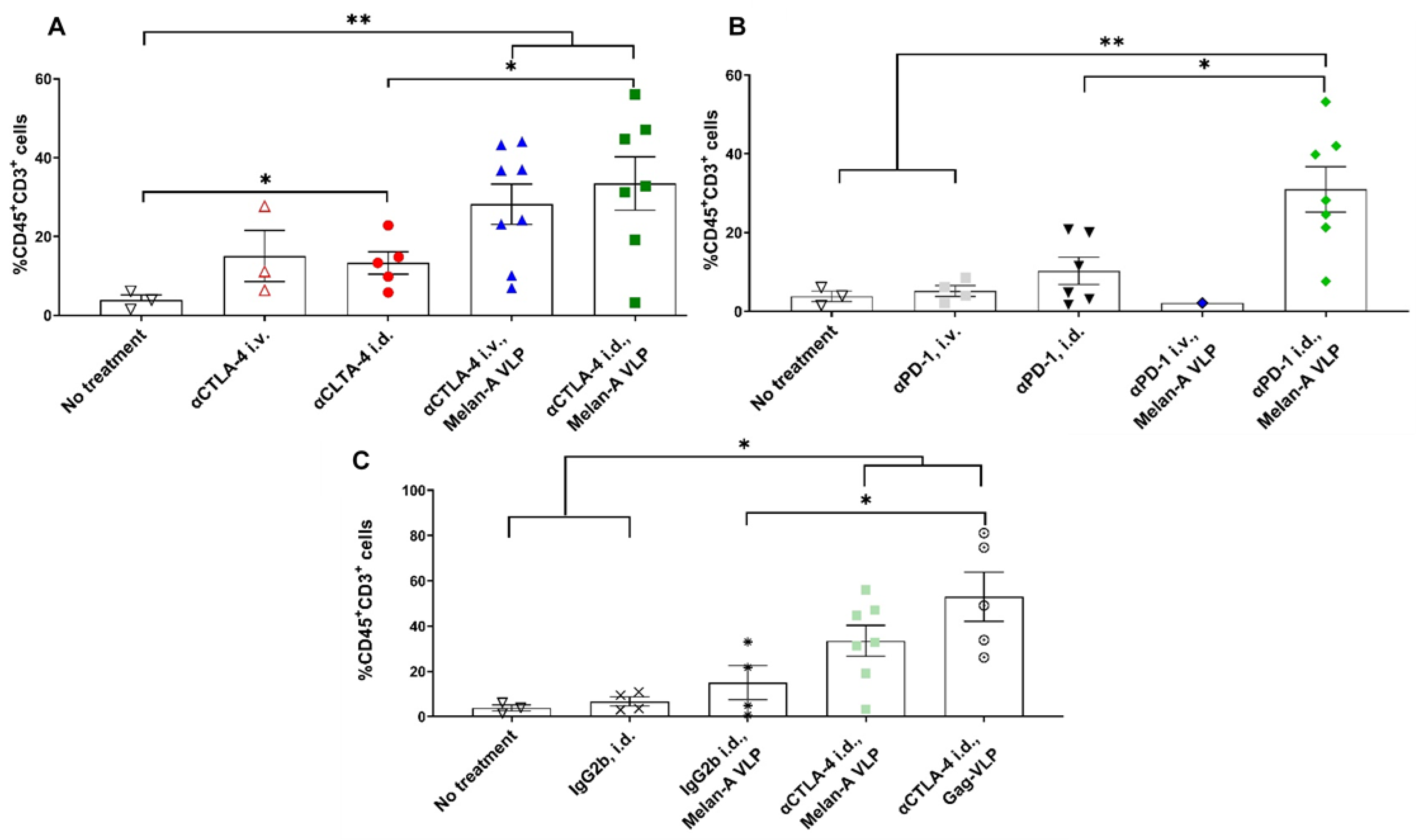
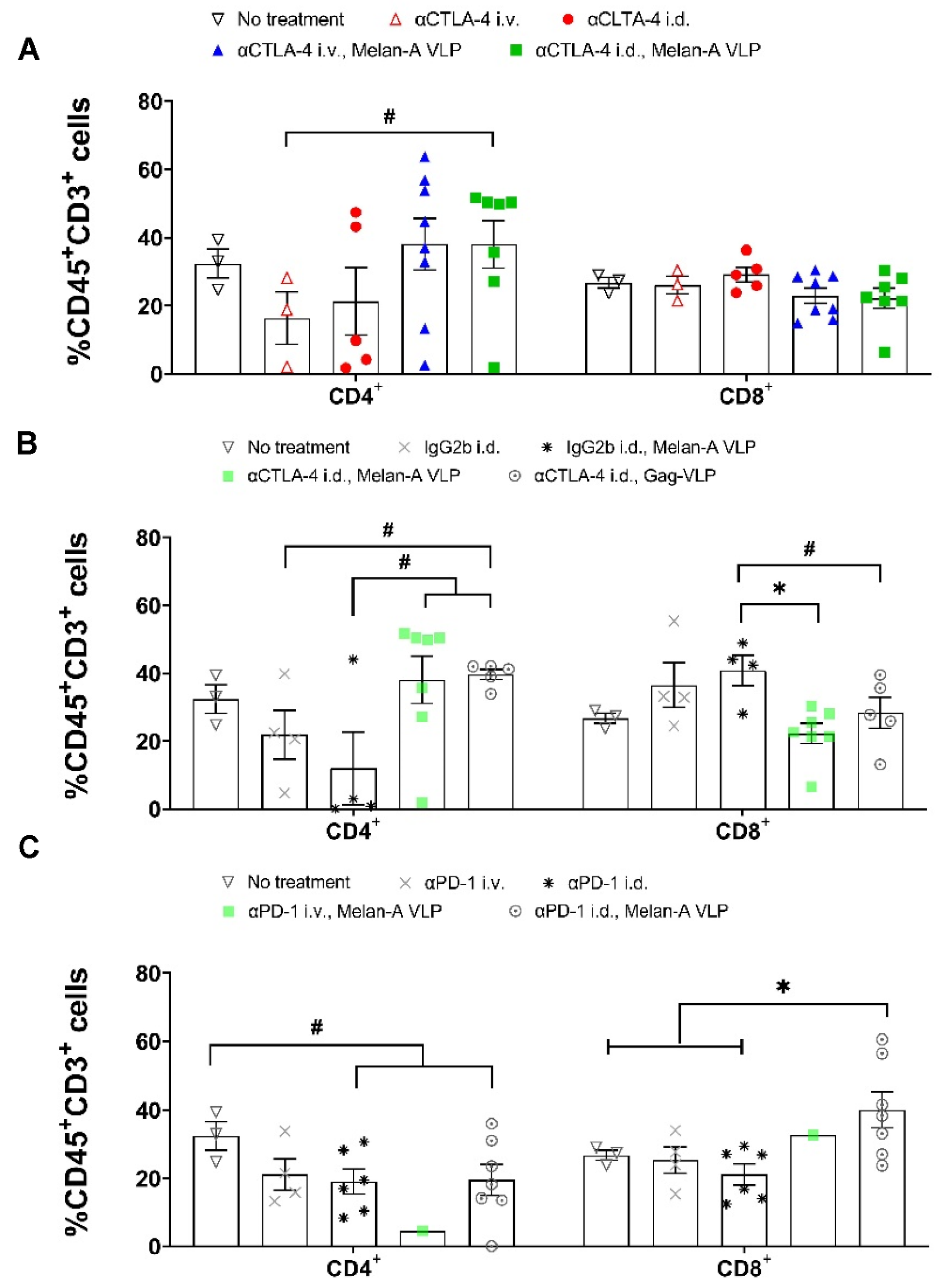
3.3. Melan-A VLP Vaccination and Lymphatic Delivery of CBI Impacts the TILs
3.4. Tumor Irrelevant Vaccination Creates a Pro-Tumor TME
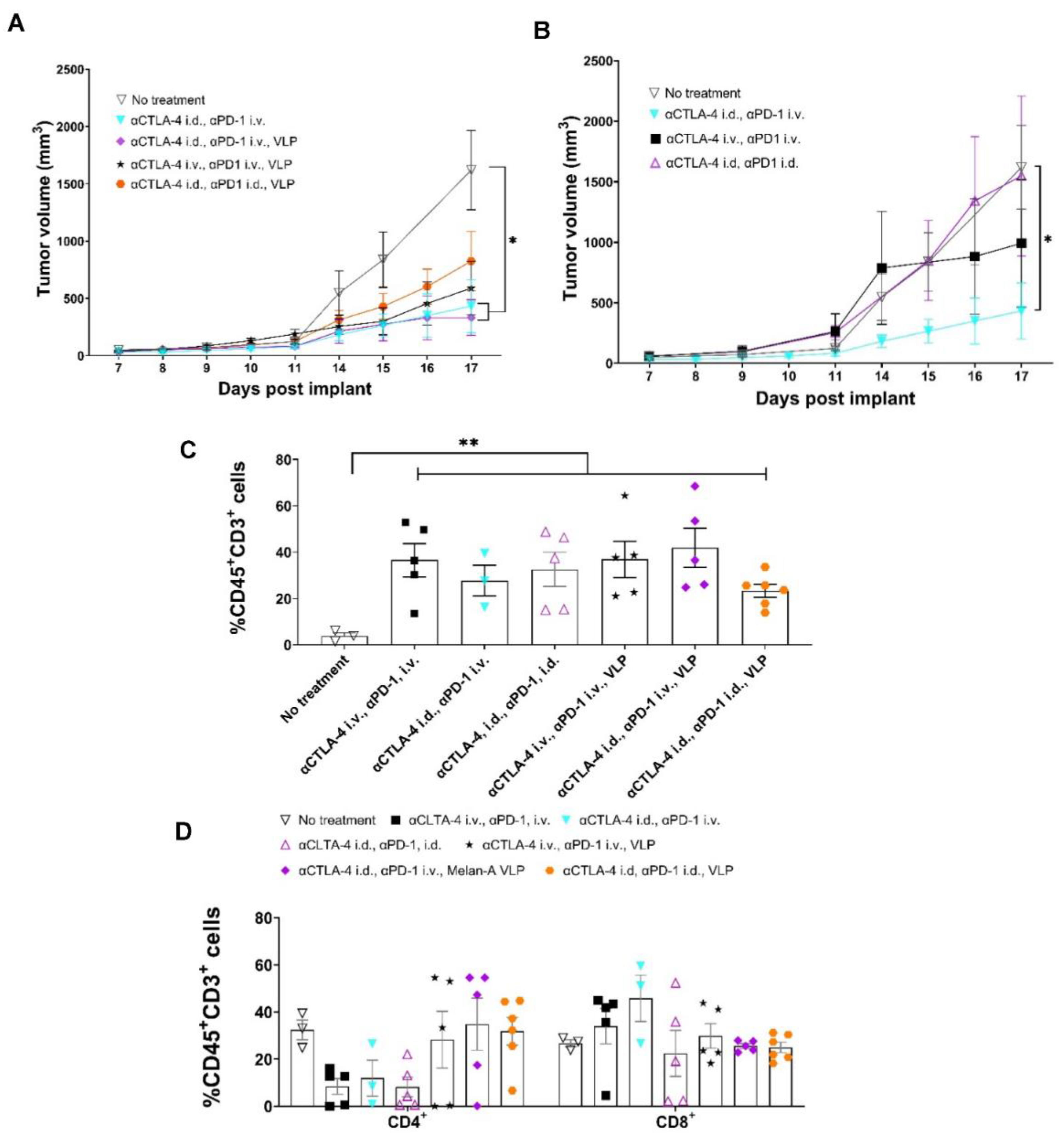
3.5. Tumor Growth Rates Significantly Reduced with i.d. Delivery of αCTLA-4 in Combination Therapy
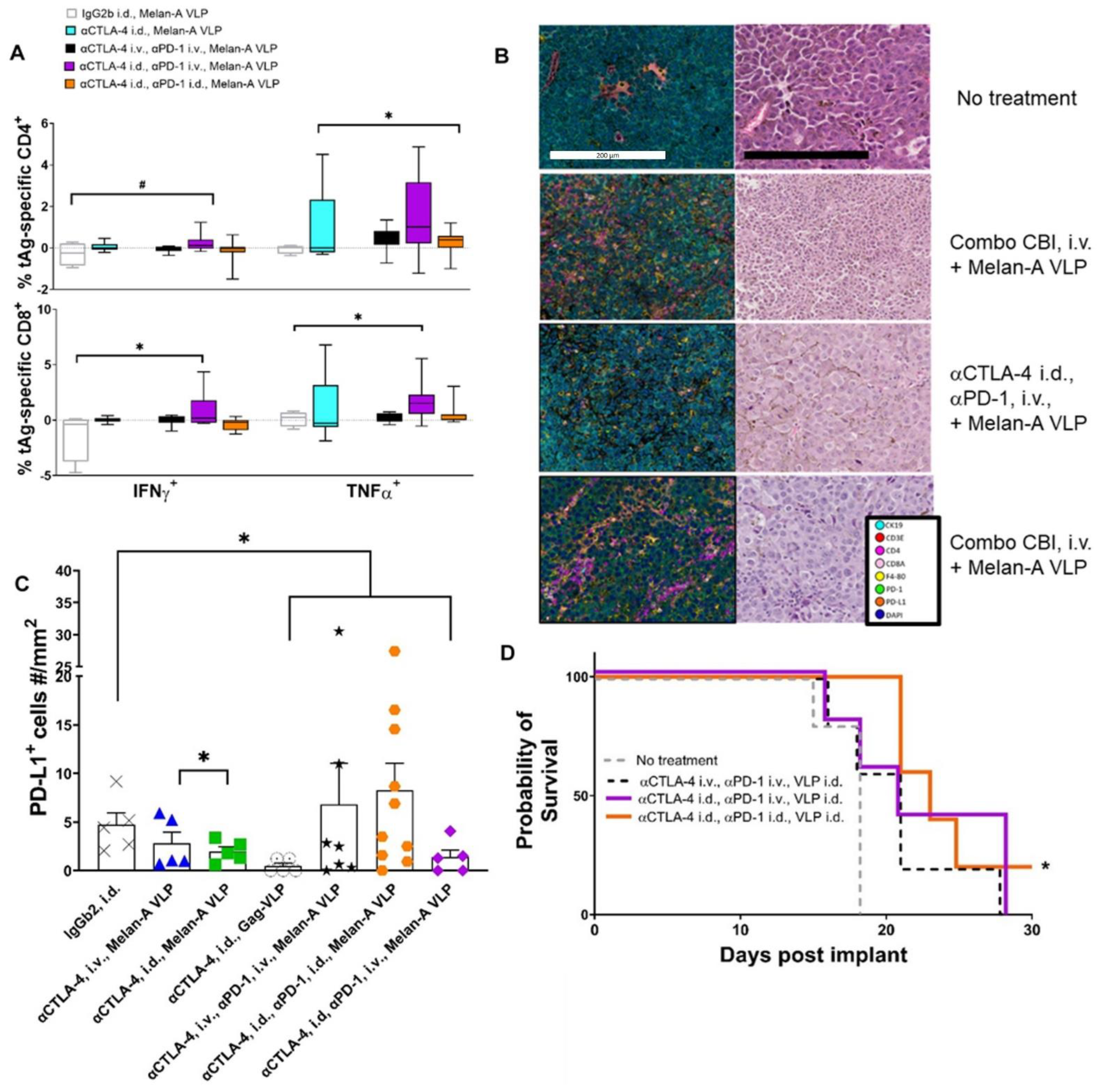
3.6. Lymphatic Delivery of CBIs Decreases Metastatic Potential in an Orthotopic Melanoma Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, L.C.; Lao, C.; Schadendorf, D.; Ferrucci, P.F.; Smylie, M.; et al. Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). In Proceedings of the American Society of Clinical Oncology 2016 Annual Meeting, Chicago, IL, USA, 3–7 June 2016. [Google Scholar]
- Sosman, J.A. Immunotherapy of Advanced Melanoma with Immune Checkpint Inhibition; UpToDate: Waltham, MA, USA, 2017. [Google Scholar]
- Metcalfe, W.; Anderson, J.; Trinh, V.A.; Hwu, W.J. Anti-programmed cell death-1 (PD-1) monoclonal antibodies in treating advanced melanoma. Discov. Med. 2015, 19, 393–401. [Google Scholar] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Carlino, M.S.; Atkinson, V.; Cebon, J.S.; Jameson, M.B.; Fitzharris, B.M.; McNeil, C.M.; Hill, A.G.; Ribas, A.; Atkins, M.B.; Thompson, J.A.; et al. KEYNOTE-029: Efficacy and Safety of Pembrolizumab Plus Ipilimumab For Advanced Melanoma. In Proceedings of the American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 1–5 June 2017. [Google Scholar]
- Weber, J.S.; Gibney, G.; Sullivan, R.J.; Sosman, J.A.; Slingluff, C.L., Jr.; Lawrence, D.P.; Logan, T.F.; Schuchter, L.M.; Nair, S.; Fecher, L.; et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016, 17, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, S.J.; Eccles, J.D.; Riccardi, P.; Peske, J.D.; Tewalt, E.F.; Cohen, J.N.; Liblau, R.; Makinen, T.; Engelhard, V.H. Roles of lymphatic endothelial cells expressing peripheral tissue antigens in CD4 T-cell tolerance induction. Nat. Commun. 2015, 6, 6771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Card, C.M.; Yu, S.S.; Swartz, M.A. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J. Clin. Investig. 2014, 124, 943–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewalt, E.F.; Cohen, J.N.; Rouhani, S.J.; Engelhard, V.H. Lymphatic endothelial cells—Key players in regulation of tolerance and immunity. Front. Immunol. 2012, 3, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Pul, K.M.; Fransen, M.F.; van de Ven, R.; de Gruijl, T.D. Immunotherapy Goes Local: The Central Role of Lymph Nodes in Driving Tumor Infiltration and Efficacy. Front. Immunol. 2021, 12, 643291. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Martinelli, R. T Lymphocyte-Endothelial Interactions: Emerging Understanding of Trafficking and Antigen-Specific Immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Hugues, S.; Dubrot, J. Shaping of Peripheral T Cell Responses by Lymphatic Endothelial Cells. Front. Immunol. 2016, 7, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.N.; Guidi, C.J.; Tewalt, E.F.; Qiao, H.; Rouhani, S.J.; Ruddell, A.; Farr, A.G.; Tung, K.S.; Engelhard, V.H. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 2010, 207, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Alitalo, K. The lymphatic vasculature in disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Aldrich, M.B.; Velasquez, F.C.; Kwon, S.; Azhdarinia, A.; Pinkston, K.; Harvey, B.R.; Chan, W.; Rasmussen, J.C.; Ross, R.F.; Fife, C.E.; et al. Lymphatic delivery of etanercept via nanotopography improves response to collagen-induced arthritis. Arthritis Res. Ther. 2017, 19, 116. [Google Scholar] [CrossRef] [Green Version]
- Haynes, J.R.; Dokken, L.; Wiley, J.A.; Cawthon, A.G.; Bigger, J.; Harmsen, A.G.; Richardson, C. Influenza-pseudotyped Gag virus-like particle vaccines provide broad protection against highly pathogenic avian influenza challenge. Vaccine 2009, 27, 530–541. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Speiser, D.E.; Knuth, A.; Bachmann, M.F. Virus-like particles for vaccination against cancer. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1579. [Google Scholar] [CrossRef] [Green Version]
- Win, S.J.; Ward, V.K.; Dunbar, P.R.; Young, S.L.; Baird, M.A. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway. Immunol. Cell Biol. 2011, 89, 681–688. [Google Scholar] [CrossRef]
- Poteet, E.; Lewis, P.; Chen, C.; Ho, S.O.; Do, T.; Chiang, S.; Labranche, C.; Montefiori, D.; Fujii, G.; Yao, Q. Toll-like receptor 3 adjuvant in combination with virus-like particles elicit a humoral response against HIV. Vaccine 2016, 34, 5886–5894. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.C.; Tan, I.C.; Marshall, M.V.; Fife, C.E.; Sevick-Muraca, E.M. Lymphatic imaging in humans with near-infrared fluorescence. Curr. Opin. Biotechnol. 2009, 20, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, J.C.; Tan, I.C.; Naqvi, S.; Aldrich, M.B.; Maus, E.A.; Blanco, A.I.; Karni, R.J.; Sevick-Muraca, E.M. Longitudinal monitoring of the head and neck lymphatics in response to surgery and radiation. Head Neck 2017, 39, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Vosjan, M.J.; Perk, L.R.; Visser, G.W.; Budde, M.; Jurek, P.; Kiefer, G.E.; van Dongen, G.A. Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat. Protoc. 2010, 5, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.R.; Jiang, M.; Solis, L.; Mino, B.; Laberiano, C.; Hernandez, S.; Gite, S.; Verma, A.; Tetzlaff, M.; Haymaker, C.; et al. Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers 2020, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Lee, L.F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.; et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [Green Version]
- Barnes, S.; Miliani de Marval, P.; Hauser, J.; Brainard, T.; Small, D.; Synott, A.; Mullin, R. A systematic evaluation of immune checkpoint inhibitos. In Proceedings of the American Association of Cancer Research Annual Meeting, Philadelphia, PA, USA, 18–22 April 2015. [Google Scholar]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Zhang, S.; Yong, L.K.; Li, D.; Cubas, R.; Chen, C.; Yao, Q. Mesothelin virus-like particle immunization controls pancreatic cancer growth through CD8+ T cell induction and reduction in the frequency of CD4+ foxp3+ ICOS− regulatory T cells. PLoS ONE 2013, 8, e68303. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Vogel, M.; Riether, C.; Muller, J.; Salatino, S.; Ternette, N.; Gomes, A.C.; Cabral-Miranda, G.; El-Turabi, A.; Ruedl, C.; et al. Targeting Mutated Plus Germline Epitopes Confers Pre-clinical Efficacy of an Instantly Formulated Cancer Nano-Vaccine. Front. Immunol. 2019, 10, 1015. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Andtbacka, R.; Harrington, K.; Melero, I.; Leidner, R.; de Baere, T.; Robert, C.; Ascierto, P.A.; Baurain, J.F.; Imperiale, M.; et al. Starting the fight in the tumor: Expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2018, 29, 2163–2174. [Google Scholar] [CrossRef]
- Aldrich, M.B.; Rasmussen, J.C.; Fife, C.E.; Shaitelman, S.F.; Sevick-Muraca, E.M. The Development and Treatment of Lymphatic Dysfunction in Cancer Patients and Survivors. Cancers 2020, 12, 2280. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, H.; Wu, T.H.; Nachman, J.; Kast, W.M. Immunodominance and tumor escape. Semin. Cancer Biol. 2002, 12, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.; Zhang, S.; Kwon, S.; Sevick-Muraca, E.M.; Li, M.; Chen, C.; Yao, Q. Virus-like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J. Immunother. 2009, 32, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Santambrogio, L.; Berendam, S.J.; Engelhard, V.H. The Antigen Processing and Presentation Machinery in Lymphatic Endothelial Cells. Front. Immunol. 2019, 10, 1033. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Velasquez, F.C.; Rasmussen, J.C.; Greives, M.R.; Turner, K.D.; Morrow, J.R.; Hwu, W.J.; Ross, R.F.; Zhang, S.; Sevick-Muraca, E.M. Nanotopography-based lymphatic delivery for improved anti-tumor responses to checkpoint blockade immunotherapy. Theranostics 2019, 9, 8332–8343. [Google Scholar] [CrossRef]
- Liu, J.; Blake, S.J.; Harjunpaa, H.; Fairfax, K.A.; Yong, M.C.; Allen, S.; Kohrt, H.E.; Takeda, K.; Smyth, M.J.; Teng, M.W. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016, 76, 5288–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amani, H.; Shahbazi, M.A.; D’Amico, C.; Fontana, F.; Abbaszadeh, S.; Santos, H.A. Microneedles for painless transdermal immunotherapeutic applications. J. Control. Release 2021, 330, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Permana, A.D.; Nainu, F.; Moffatt, K.; Larraneta, E.; Donnelly, R.F. Recent advances in combination of microneedles and nanomedicines for lymphatic targeted drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1690. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantilla-Rojas, C.; Velasquez, F.C.; Morton, J.E.; Clemente, L.C.; Parra, E.R.; Torres-Cabala, C.; Sevick-Muraca, E.M. Enhanced T-Cell Priming and Improved Anti-Tumor Immunity through Lymphatic Delivery of Checkpoint Blockade Immunotherapy. Cancers 2022, 14, 1823. https://doi.org/10.3390/cancers14071823
Mantilla-Rojas C, Velasquez FC, Morton JE, Clemente LC, Parra ER, Torres-Cabala C, Sevick-Muraca EM. Enhanced T-Cell Priming and Improved Anti-Tumor Immunity through Lymphatic Delivery of Checkpoint Blockade Immunotherapy. Cancers. 2022; 14(7):1823. https://doi.org/10.3390/cancers14071823
Chicago/Turabian StyleMantilla-Rojas, Carolina, Fred C. Velasquez, Janelle E. Morton, Leticia C. Clemente, Edwin R. Parra, Carlos Torres-Cabala, and Eva M. Sevick-Muraca. 2022. "Enhanced T-Cell Priming and Improved Anti-Tumor Immunity through Lymphatic Delivery of Checkpoint Blockade Immunotherapy" Cancers 14, no. 7: 1823. https://doi.org/10.3390/cancers14071823