
An Immunocompetent Environment Unravels the Proto-Oncogenic Role of miR-22
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Breast Cancer
3. Prostate Cancer
4. Leukemia
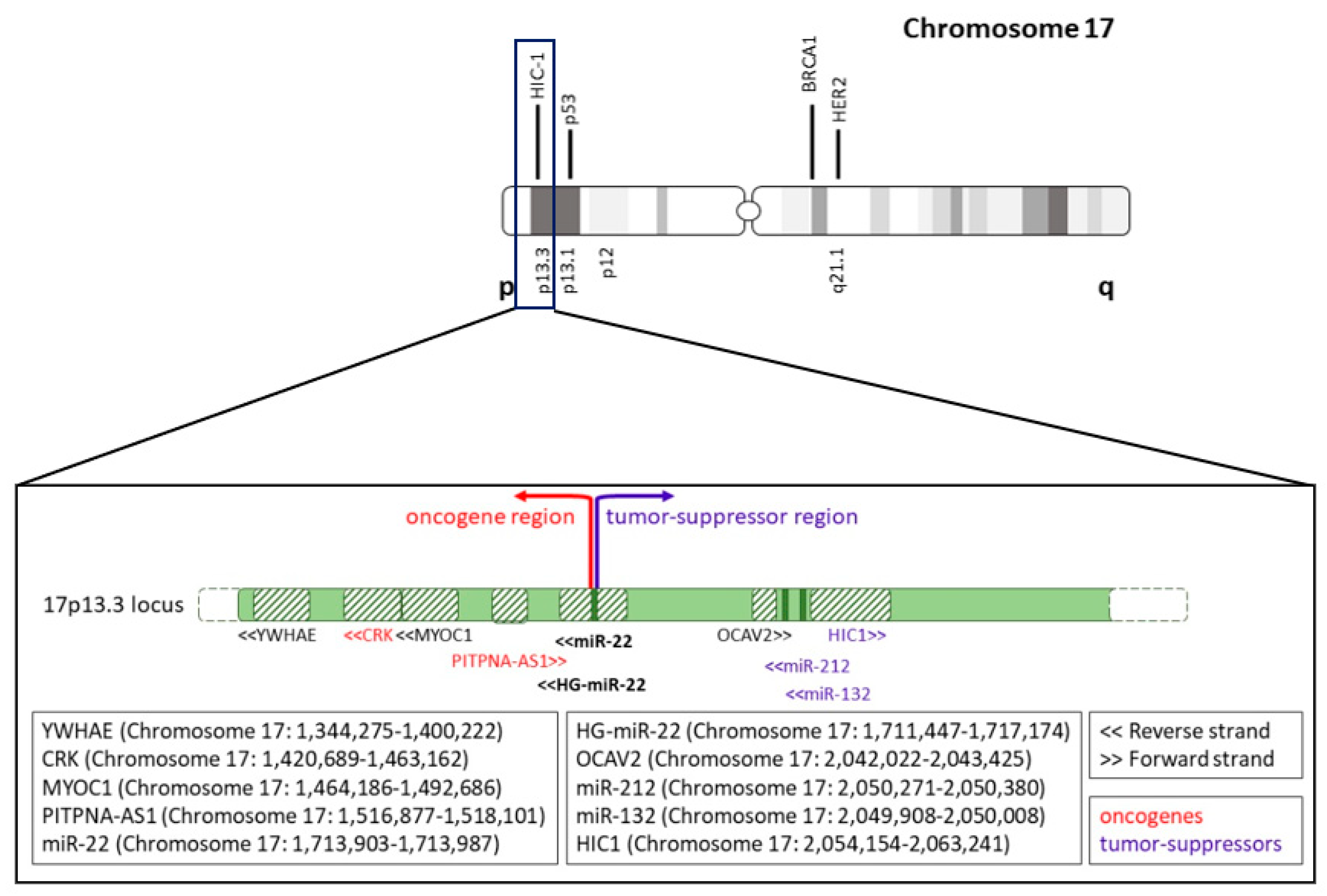
5. Alterations of miR-22 Gene
6. Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Ankasha, S.J.; Shafiee, M.N.; Wahab, N.A.; Ali, R.A.R.; Mokhtar, N.M. Post-transcriptional regulation of microRNAs in cancer: From prediction to validation. Oncol. Rev. 2018, 12, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Shyr, Y.; Cai, J.; Liu, Q. Interplay between miRNAs and host genes and their role in cancer. Brief. Funct. Genom. 2018, 18, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Liu, M.; Cao, Y. New Insight into microRNA Functions in Cancer: Oncogene-microRNA-Tumor Suppressor Gene Network. Front. Mol. Biosci. 2017, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, M.; Tuccoli, A.; D’Aurizio, R.; Sarti, S.; Giannecchini, L.; Lubrano, S.; Marranci, A.; Evangelista, M.; Peppicelli, S.; Ippolito, C.; et al. Context-dependent miR-204 and miR-211 affect the biological properties of amelanotic and melanotic melanoma cells. Oncotarget 2017, 8, 25395–25417. [Google Scholar] [CrossRef] [Green Version]
- Nejati, K.; Alivand, M.R.; Arabzadeh, A.A. MicroRNA-22 in female malignancies: Focusing on breast, cervical, and ovarian cancers. Pathol. Res. Pract. 2021, 223. [Google Scholar] [CrossRef]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic MicroRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.J.; Pandolfi, P.P. miR-22 in tumorigenesis. Cell Cycle 2014, 13, 11–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurm, A.A.; Tenen, D.G.; Behre, G. The Janus-faced Nature of miR-22 in Hematopoiesis: Is It an Oncogenic Tumor Suppressor or Rather a Tumor-Suppressive Oncogene? PLoS Genet. 2017, 13, e1006505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Li, X.Y.; Wang, Z.M.; Han, Z.F.; Zhao, Y.H. MiR-22 inhibits lung cancer cell EMT and invasion through targeting Snail. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3598–3604. [Google Scholar]
- Chen, M.; Hu, W.; Xiong, C.L.; Qu, Z.; Yin, C.Q.; Wang, Y.H.; Luo, C.L.; Guan, Q.; Yuan, C.H.; Wang, F.B. miR-22 targets YWHAZ to inhibit metastasis of hepatocellular carcinoma and its down-regulation predicts a poor survival. Oncotarget 2016, 7, 80751–80764. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; He, M.; Wan, D.; Ye, Y.; He, Y.; Han, L.; Guo, M.; Huang, Y.; Qin, W.; Wang, M.W.; et al. The minimum LOH region defined on chromosome 17p13.3 in human hepatocellular carcinoma with gene content analysis. Cancer Lett. 2003, 190, 221–232. [Google Scholar] [CrossRef]
- Quintana, P.J.; Neuwirth, E.A.; Grosovsky, A.J. Interchromosomal gene conversion at an endogenous human cell locus. Genetics 2001, 158, 757–767. [Google Scholar] [CrossRef]
- Huang, Z.P.; Wang, D.Z. miR-22 in cardiac remodeling and disease. Trends Cardiovasc. Med. 2014, 24, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Rastegar-moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. MicroRNA-22: A Novel and Potent Biological Therapeutics in Neurological Disorders. Mol. Neurobiol. 2022, 59, 2694–2701. [Google Scholar] [CrossRef]
- Zhang, C.; Fang, L.; Liu, X.; Nie, T.; Li, R.; Cui, L.; Wang, J.; Ji, Y. miR-22 inhibits synovial fibroblasts proliferation and proinflammatory cytokine production in RASF via targeting SIRT1. Gene 2020, 724, 144144. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Yang, K.P.; Li, Z.Z. MiR-22 restrains proliferation of rheumatoid arthritis by targeting IL6R and may be concerned with the suppression of NF-κB pathway. Kaohsiung J. Med. Sci. 2020, 36, 20–26. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.S.; Mugiyanto, E.; Chang, W.C.; Wan, Y.J.Y. MiR-22 as a metabolic silencer and liver tumor suppressor. Liver Res. 2020, 4, 74–80. [Google Scholar] [CrossRef]
- Wang, B.; Yao, Q.; Xu, D.; Zhang, J.A. MicroRNA-22-3p as a novel regulator and therapeutic target for autoimmune diseases. Int. Rev. Immunol. 2017, 36, 176–181. [Google Scholar] [CrossRef]
- Diniz, G.P.; Huang, Z.P.; Liu, J.; Chen, J.; Ding, J.; Fonseca, R.I.; Barreto-Chaves, M.L.; Donato, J.; Hu, X.; Wang, D.Z. Loss of microRNA-22 prevents high-fat diet induced dyslipidemia and increases energy expenditure without affecting cardiac hypertrophy. Clin. Sci. 2017, 131, 2885–2900. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.J.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. Innov. Hepatol. 2020, 2, 100093. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.F.; Cao, L.Y.; Yu, T.; Gong, L.; Wang, L.N.; Zhao, Y.L.; Xiao, B.; Zou, Q.M. MicroRNA-22 inhibits tumor growth and metastasis in gastric cancer by directly targeting MMP14 and Snail. Cell Death Dis. 2015, 6, e2000. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Kumar, A.; Gomez, C.R.; Akhtar, I.; Hancock, J.C.; Lage, J.M.; Pound, C.R.; Levenson, A.S. MTA1-activated Epi-microRNA-22 regulates E-cadherin and prostate cancer invasiveness. FEBS Lett. 2017, 591, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Wang, D.; Wei, X. miR-22 suppresses tumorigenesis and improves radiosensitivity of breast cancer cells by targeting Sirt1. Biol. Res. 2017, 50, 27. [Google Scholar] [CrossRef]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Hu, C.; Arnovitz, S.; Bugno, J.; Yu, M.; Zuo, Z.; Chen, P.; Huang, H.; Ulrich, B.; Gurbuxani, S.; et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat. Commun. 2016, 7, 11452. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Zhang, P.; Wen, L.; Jia, S.; Wu, Y.; Zhang, Z.; Guan, L.; Yu, Z.; Zhao, L. miR-22 promotes stem cell traits via activating Wnt/β-catenin signaling in cutaneous squamous cell carcinoma. Oncogene 2021, 40, 5799–5813. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Yang, T.; Liu, Y.; Li, A.; Fu, S.; Wu, M.; Pan, Z.; Zhou, W. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br. J. Cancer 2010, 103, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Parrish, J.K.; Sechler, M.; Winn, R.A.; Jedlicka, P. The histone demethylase KDM3A is a microRNA-22-regulated tumor promoter in Ewing Sarcoma. Oncogene 2015, 34, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, L.; Wang, T.; Li, Y.; Xun, Q.; Zhang, R.; Liu, L.; Li, L.; Wang, W.; Tian, Y.; et al. Tumor cell-secreted exosomal miR-22-3p inhibits transgelin and induces vascular abnormalization to promote tumor budding. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 2151–2166. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; van Diest, P.; Raman, V. Twist activates miR-22 to suppress estrogen receptor alpha in breast cancer. Mol. Cell. Biochem. 2021, 476, 2295–2306. [Google Scholar] [CrossRef]
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518. [Google Scholar] [CrossRef]
- Pandey, A.K.; Zhang, Y.; Zhang, S.; Li, Y.; Tucker-Kellogg, G.; Yang, H.; Jha, S. TIP60-miR-22 axis as a prognostic marker of breast cancer progression. Oncotarget 2015, 6, 41290–41306. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, D.; Filkowski, J.; Rodriguez-Juarez, R.; Storozynsky, Q.; Malach, M.; Carpenter, E.; Kovalchuk, O. A dual role of miR-22 modulated by RelA/p65 in resensitizing fulvestrant-resistant breast cancer cells to fulvestrant by targeting FOXP1 and HDAC4 and constitutive acetylation of p53 at Lys382. Oncogenesis 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Gorur, A.; Bayraktar, R.; Ivan, C.; Mokhlis, H.A.; Bayraktar, E.; Kahraman, N.; Karakas, D.; Karamil, S.; Kabil, N.N.; Kanlikilicer, P.; et al. ncRNA therapy with miRNA-22-3p suppresses the growth of triple-negative breast cancer. Mol. Ther. Nucleic Acids 2021, 23, 930–943. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.M.; Liao, C.G.; Zhang, Y.; Xu, J.; Li, Y.; Huang, W.; Bian, H.; Chen, Z.N. A regulatory loop involving miR-22, Sp1, and c-Myc modulates CD147 expression in breast cancer invasion and metastasis. Cancer Res. 2014, 74, 3764–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Wang, J.; He, Q.; Liu, L.; Wang, Z. AC016405.3 functions as an oncogenic long non-coding RNA by regulating ERBB3 via sponging miR-22-3p in breast cancer. J. Clin. Lab. Anal. 2021, 35, e23952. [Google Scholar] [CrossRef]
- Che, L.; Yang, H.; Wang, D.; Liu, S. Corylin sensitizes breast cancer cells to overcome tamoxifen resistance by regulating OAS1/miR-22-3p/SIRT1 axis. Acta Biochim. Pol. 2021, 68, 757–764. [Google Scholar] [CrossRef]
- Fan, T.; Wang, C.Q.; Li, X.T.; Yang, H.; Zhou, J.; Song, Y.J. MiR-22-3p Suppresses Cell Migration and Invasion by Targeting PLAGL2 in Breast Cancer. J. Coll. Physicians Surg.—Pak. JCPSP 2021, 31, 937–940. [Google Scholar] [CrossRef]
- Wang, X.; Yao, Z.; Fang, L. miR-22-3p/PGC1beta Suppresses Breast Cancer Cell Tumorigenesis via PPARgamma. PPAR Res. 2021, 2021, 6661828. [Google Scholar] [CrossRef]
- Pandey, D.P.; Picard, D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell. Biol. 2009, 29, 3783–3790. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.K.; Wang, Y.; Wen, Y.Y.; Zhao, P.; Bian, Z.J. MicroRNA-22 Suppresses Breast Cancer Cell Growth and Increases Paclitaxel Sensitivity by Targeting NRAS. Technol. Cancer Res. Treat. 2018, 17, 1533033818809997. [Google Scholar] [CrossRef] [Green Version]
- Shao, P.; Liu, Q.; Maina, P.K.; Cui, J.; Bair, T.B.; Li, T.; Umesalma, S.; Zhang, W.; Qi, H.H. Histone demethylase PHF8 promotes epithelial to mesenchymal transition and breast tumorigenesis. Nucleic Acids Res. 2017, 45, 1687–1702. [Google Scholar] [CrossRef]
- Xiong, J.; Yu, D.; Wei, N.; Fu, H.; Cai, T.; Huang, Y.; Wu, C.; Zheng, X.; Du, Q.; Lin, D.; et al. An estrogen receptor alpha suppressor, microRNA-22, is downregulated in estrogen receptor alpha-positive human breast cancer cell lines and clinical samples. FEBS J. 2010, 277, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.B.; Appaiah, H.N.; Burnett, R.M.; Bhat-Nakshatri, P.; Wang, G.; Mehta, R.; Badve, S.; Thomson, M.J.; Hammond, S.; Steeg, P.; et al. Control of EVI-1 oncogene expression in metastatic breast cancer cells through microRNA miR-22. Oncogene 2011, 30, 1290–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damavandi, Z.; Torkashvand, S.; Vasei, M.; Soltani, B.M.; Tavallaei, M.; Mowla, S.J. Aberrant Expression of Breast Development-Related MicroRNAs, miR-22, miR-132, and miR-212, in Breast Tumor Tissues. J. Breast Cancer 2016, 19, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Tang, H.; Liu, X.; Liu, P.; Yang, L.; Xie, X.; Ye, F.; Song, C.; Wei, W. miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015, 356, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Hemani, R.; Patel, I.; Inamdar, N.; Campanelli, G.; Donovan, V.; Kumar, A.; Levenson, A.S. Dietary Pterostilbene for MTA1-Targeted Interception in High-Risk Premalignant Prostate Cancer. Cancer Prev. Res. 2022, 15, 87–100. [Google Scholar] [CrossRef]
- Lin, Y.; Miao, Z.; Zhang, X.; Wei, X.; Hou, J.; Huang, Y.; Shen, B. Identification of Key MicroRNAs and Mechanisms in Prostate Cancer Evolution Based on Biomarker Prioritization Model and Carcinogenic Survey. Front. Genet. 2020, 11, 596826. [Google Scholar] [CrossRef]
- Joshi, T.; Patel, I.; Kumar, A.; Donovan, V.; Levenson, A.S. Grape Powder Supplementation Attenuates Prostate Neoplasia Associated with Pten Haploinsufficiency in Mice Fed High-Fat Diet. Mol. Nutr. Food Res. 2020, 64, e2000326. [Google Scholar] [CrossRef]
- Deng, Z.H.; Yu, G.S.; Pan, B.; Feng, Z.H.; Huang, Q.; Deng, J.Z.; Chen, B.; Yang, S.K. Rs145204276 and rs4759314 affect the prognosis of prostate cancer by modulating the GAS5/miR-1284/HMGB1 and HOTAIR/miR-22/HMGB1 signalling pathways. Artif. Cells Nanomed. Biotechnol. 2020, 48, 435–442. [Google Scholar] [CrossRef]
- Knyazev, E.N.; Samatov, T.R.; Fomicheva, K.A.; Nyushko, K.M.; Alekseev, B.Y.; Shkurnikov, M.Y. MicroRNA hsa-miR-4674 in Hemolysis-Free Blood Plasma Is Associated with Distant Metastases of Prostatic Cancer. Bull. Exp. Biol. Med. 2016, 161, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Budd, W.T.; Seashols-Williams, S.J.; Clark, G.C.; Weaver, D.; Calvert, V.; Petricoin, E.; Dragoescu, E.A.; O’Hanlon, K.; Zehner, Z.E. Dual Action of miR-125b As a Tumor Suppressor and OncomiR-22 Promotes Prostate Cancer Tumorigenesis. PLoS ONE 2015, 10, e0142373. [Google Scholar] [CrossRef] [Green Version]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualini, L.; Bu, H.; Puhr, M.; Narisu, N.; Rainer, J.; Schlick, B.; Schafer, G.; Angelova, M.; Trajanoski, Z.; Borno, S.T.; et al. miR-22 and miR-29a Are Members of the Androgen Receptor Cistrome Modulating LAMC1 and Mcl-1 in Prostate Cancer. Mol. Endocrinol. 2015, 29, 1037–1054. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kantarjian, H.; Guo, Y.; Lin, E.; Shan, J.; Huang, X.; Berry, D.; Ahmed, S.; Zhu, W.; Pierce, S.; et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Pulikkan, J.A.; Peramangalam, P.S.; Dengler, V.; Ho, P.A.; Preudhomme, C.; Meshinchi, S.; Christopeit, M.; Nibourel, O.; Müller-Tidow, C.; Bohlander, S.K.; et al. C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood 2010, 116, 5638–5649. [Google Scholar] [CrossRef] [Green Version]
- Katzerke, C.; Madan, V.; Gerloff, D.; Bräuer-Hartmann, D.; Hartmann, J.U.; Wurm, A.A.; Müller-Tidow, C.; Schnittger, S.; Tenen, D.G.; Niederwieser, D.; et al. Transcription factor C/EBPα-induced microRNA-30c inactivates Notch1 during granulopoiesis and is downregulated in acute myeloid leukemia. Blood 2013, 122, 2433–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucci, G.; Maharry, K.S.; Metzeler, K.H.; Volinia, S.; Wu, Y.Z.; Mrozek, K.; Nicolet, D.; Kohlschmidt, J.; Whitman, S.P.; Mendler, J.H.; et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2086–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Lu, J.; Sun, M.; Li, Z.; Zhang, H.; Neilly, M.B.; Wang, Y.; Qian, Z.; Jin, J.; Zhang, Y.; et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 19971–19976. [Google Scholar] [CrossRef] [PubMed]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Patnaik, M.M.; Hanson, C.A.; Pardanani, A.; Gilliland, D.G.; Levine, R.L. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009, 23, 1343–1345. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Chen, M.T.; Zhang, X.H.; Yin, X.L.; Ning, H.M.; Su, R.; Lin, H.S.; Song, L.; Wang, F.; Ma, Y.N.; et al. The PU.1-Modulated MicroRNA-22 Is a Regulator of Monocyte/Macrophage Differentiation and Acute Myeloid Leukemia. PLoS Genet. 2016, 12, e1006259. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, S.; Tyybäkinoja, A.; Borze, I.; Räty, R.; Saarinen-Pihkala, U.M.; Usvasalo, A.; Elonen, E.; Knuutila, S. Integrated analysis of gene copy number, copy neutral LOH, and microRNA profiles in adult acute lymphoblastic leukemia. Cytogenet. Genome Res. 2012, 136, 246–255. [Google Scholar] [CrossRef]
- Kashiwagi, H.; Uchida, K. Genome-wide profiling of gene amplification and deletion in cancer. Hum. Cell 2000, 13, 135–141. [Google Scholar]
- Park, T. Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells 2021, 10, 739. [Google Scholar] [CrossRef]
- Liu, L.; Dai, A.; Zhang, Z.; Ning, M.; Han, D.; Li, L.; Li, Z. LncRNA PITPNA-AS1 promotes gastric cancer by increasing SOX4 expression via inhibition of miR-92a-3p. Aging 2021, 13, 21191–21201. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, L.; Bo, Q.; Chen, L.; Sun, L.; Shi, H. Long noncoding RNA PITPNA-AS1 promotes cervical cancer progression through regulating the cell cycle and apoptosis by targeting the miR-876-5p/c-MET axis. Biomed. Pharmacother.=Biomed. Pharmacother. 2020, 128, 110072. [Google Scholar] [CrossRef]
- Yuan, C.; Yang, L. Long Non-Coding RNA PITPNA-AS1 Accelerates the Progression of Colorectal Cancer Through miR-129-5p/HMGB1 Axis. Cancer Manag. Res. 2020, 12, 12497–12507. [Google Scholar] [CrossRef] [PubMed]
- Incoronato, M.; Urso, L.; Portela, A.; Laukkanen, M.O.; Soini, Y.; Quintavalle, C.; Keller, S.; Esteller, M.; Condorelli, G. Epigenetic regulation of miR-212 expression in lung cancer. PLoS ONE 2011, 6, e27722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Ou, Y.L.; Huang, Z.P.; Hong, Y.G.; Tao, Y.P.; Wang, Z.G.; Ni, J.S.; Hao, L.Q.; Lin, H. MicroRNA-212-3p inhibits the Proliferation and Invasion of Human Hepatocellular Carcinoma Cells by Suppressing CTGF expression. Sci. Rep. 2019, 9, 9820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Ji, Z.; Yan, W.; Zhou, Z.; Li, H. The biological functions and mechanism of miR212 in prostate cancer proliferation, migration and invasion via targeting Engrailed-2. Oncol. Rep. 2017, 38, 1411–1419. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, C.; Shen, C.; Yin, F.; Wang, K.; Liu, Y.; Zheng, B.; Zhang, W.; Hou, X.; Chen, X.; et al. MiR-212-3p inhibits glioblastoma cell proliferation by targeting SGK3. J. Neuro-Oncol. 2015, 122, 431–439. [Google Scholar] [CrossRef]
- Moghbeli, M.; Zangouei, A.S.; Nasrpour Navaii, Z.; Taghehchian, N. Molecular mechanisms of the microRNA-132 during tumor progressions. Cancer Cell Int. 2021, 21, 439. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Y.; Ling, Y.; Zhou, C.; Wang, H.; Teschendorff, A.E.; Zhao, Y.; Zhao, H.; He, Y.; Zhang, G.; et al. dbDEMC 3.0: Functional exploration of differentially expressed miRNAs in cancers of human and model organisms. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Nam, J.W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Almeida Silva, L.F.; Reschke, C.R.; Nguyen, N.T.; Langa, E.; Sanz-Rodriguez, A.; Gerbatin, R.R.; Temp, F.R.; de Freitas, M.L.; Conroy, R.M.; Brennan, G.P.; et al. Genetic deletion of microRNA-22 blunts the inflammatory transcriptional response to status epilepticus and exacerbates epilepsy in mice. Mol. Brain 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; You, R.; Yuan, X.; Yang, T.; Samuel, E.L.; Marcano, D.C.; Sikkema, W.K.; Tour, J.M.; Rodriguez, A.; Kheradmand, F.; et al. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat. Immunol. 2015, 16, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Liu, Y.; Mei, S.; Zhang, M.; Xin, J.; Zhang, Y.; Yang, R. MicroRNA-22 impairs anti-tumor ability of dendritic cells by targeting p38. PLoS ONE 2015, 10, e0121510. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunosuppression and autoimmune disease: Beyond immunosuppressive networks for tumour immunity. Immunology 2006, 119, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Qiu, R.; Zhang, Z.; Han, Z.; Yao, C.; Hou, G.; Dai, D.; Jin, W.; Tang, Y.; Yu, X.; et al. The MicroRNA miR-22 Represses Th17 Cell Pathogenicity by Targeting PTEN-Regulated Pathways. ImmunoHorizons 2020, 4, 308–318. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target Gene | Tumor | Pathway | Reference |
|---|---|---|---|
| SIRT1 | breast, ovarian, cervical, HCC | pRB | 21502362 (Xu, D., 2011); 28882183 (Zhang, X., 2017) |
| CDK6 | breast | pRB | 21502362 (Xu, D., 2011) |
| CD147 | breast | miR-22/Sp1/c-Myc | 24906624 (Kong, L.M., 2014) |
| ERa | breast | PKC/ERK | 19414598 (Pandey, D.P., 2009); 33173749 (Gao, Y., 2020) |
| Erbb3 | lung | PI3K/AKT | 22484852 (Ling, B., 2012) |
| WNT1 | GC | Wnt/β-catenin | 23851184 (Tang, H., 2013) |
| CD151 | GC | - | 24495805 (Wang, X., 2014) |
| CDKN1A | HCC | - | 23582783 (Shi, C., 2013) |
| NCOA1 | HCC | NF/kB | 21798241 (Takara, A., 2011) |
| GLUT1 | breast | - | 25304371 (Chen, B., 2015) |
| SP1 | breast, cervical, GC, HCC | pRB | 21502362 (Xu, D., 2011); 23529765 (Guo, M.M., 2013). |
| HDAC4 | HCC | - | 20842113 (Zhang, J., 2010) 23349832 (Jovicic, A., 2013) |
| MYCBP | breast | MYCBP/c-Myc | 20562918 (Xiong, J. 2010) |
| HDAC6 | cervical | E6/p53 | 30379969 (Wongjampa, W. 2018) |
| LGALS1 | RCC | HIF/mTOR | 24496460 (White, N.M., 2014) |
| HIF1A | RCC | HIF/mTOR | 24496460 (White, N.M., 2014) |
| NET1 | CML | Net1/RhoA | 25041463 (Ahmad, H.M., 2014) |
| TIAM1 | CRC | Rac/Rho | 23440286 (Li, B., 2013) |
| BTG1 | CRC | BTG1/LC3II; apoptosis | 25449431 (Zhang, H., 2015) |
| LGALS9 | HCC | Tim3/Gal-9 | 26239725 (Yang, Q., 2015) |
| MMP14 | GC | ECM, EMT | 26610210 (Zuo, Q.F., 2015) |
| SNAIL | GC | ECM, EMT | 26610210 (Zuo, Q.F., 2015) |
| ACLY | osteosarcoma, prostate, cervical, lung, breast | FASN/HMGCR | 27317765 (Xin, M., 2016); 29636857 (Liu, H., 2018) |
| TIP60 * | breast | EMT | 26512777 (Pandey, A.K., 2015) |
| NRAS * | breast | PI3K/AKT, MAPK/ERK, NF-kB | 30384806 (Song, Y.K., 2018) |
| TWIST1 * | osteosarcoma | EMT | 32391253 (Zhu, S.T., 2020) |
| TET2 | MDS, breast, HCC | Epigenetic | 23827711 (Song, S.J., 2013) 23830207 (Song, S.J., 2013) 34019487 (Chen, D., 2021) |
| PTEN | prostate, breast | PI3K/Akt | 20388916 (Poliseno, L., 2010) 33173749 (Gao, Y., 2020) |
| CBL * | papillary thyroid | Wnt/β-catenin | 30190130 (Wang, M., 2018) |
| FOSB * | cSCC | Wnt/β-catenin | 34345013 (Yuan, S., 2021) |
| PAD2 * | cSCC | Wnt/β-catenin | 34345013 (Yuan, S., 2021) |
| Tumor | miR-22 Role | Xenograft | Trangenic | Knock-Out | Reference |
|---|---|---|---|---|---|
| AML | tumor suppressor | ✔ | Shen, C. et al., 2016 | ||
| tumor suppressor | ✔ | ✔ * | Jiang, X. et al., 2016 | ||
| Breast Cancer | oncogene | ✔ | ✔ * | Gao et al., 2020 | |
| oncogene | ✔ | ✔ | Song et al., 2013 | ||
| tumor suppressor | ✔ | Gorur, A. et al., 2021 | |||
| tumor suppressor | ✔ | Liu, X. et al., 2018 | |||
| tumor suppressor | ✔ | Liu, H. et al., 2018 | |||
| tumor suppressor | ✔ | Shao, P. et al., 2017 | |||
| tumor suppressor | ✔ | Kong, L.M. et al., 2014 | |||
| tumor suppressor | ✔ | Xu, D. et al., 2011 | |||
| Colon Cancer | tumor suppressor | ✔ | Cong, J. et al., 2020 | ||
| tumor suppressor | ✔ | Sun, R. et al., 2019 | |||
| tumor suppressor | ✔ | Hu, Y., 2019 | |||
| tumor suppressor | ✔ | Liu Y., 2018 | |||
| tumor suppressor | ✔ | Xia, S.S. et al., 2017 | |||
| tumor suppressor | ✔ | Zhang, H. et al., 2015 | |||
| cSCC | oncogene | ✔ | ✔ * | Yuan, S. et al., 2021 | |
| gastric cancer | tumor suppressor | ✔ | Zong, W. et al., 2020 | ||
| tumor suppressor | ✔ | Li, X. et al., 2020 | |||
| tumor suppressor | ✔ | Gan, L. et al., 2019 | |||
| tumor suppressor | ✔ | Li, S. et al., 2018 | |||
| tumor suppressor | ✔ | Tang, H. et al., 2013 | |||
| Hepatocellular Carcinoma | oncogene | ✔ | Chen, D. et al., 2021 | ||
| tumor-suppressor | ✔ | Zhang, L. et al., 2021 | |||
| tumor suppressor | ✔ | Chen, F., 2020 | |||
| tumor suppressor | ✔ | Chen, S. et al., 2020 | |||
| tumor suppressor | ✔ | Leung, Z. et al., 2019 | |||
| tumor-suppressor | ✔ | Zhao, L. et al., 2019 | |||
| tumor suppressor | ✔ | Chen, S. et al., 2018 | |||
| tumor-suppressor | ✔ | Yang, F. et al., 2016 | |||
| tumor-suppressor | ✔ | Zhang, J. et al., 2010 | |||
| MDS | oncogene | ✔ | Song, S.J. et al., 2013 | ||
| Osteosarcoma | tumor suppressor | ✔ | Xue, Y. et al., 2021 | ||
| tumor-suppressor | ✔ | Meng, C.Y. et al., 2020 | |||
| tumor-suppressor | ✔ | Meng, C.Y. et al., 2020 | |||
| tumor-suppressor | ✔ | Zhu, H. et al., 2020 | |||
| Prostate Cancer | oncogene | ✔ | Hemani, R. et al., 2022 | ||
| oncogene | ✔ | Joshi, T. et al., 2020 | |||
| oncogene | ✔ | Dhar, S. et al., 2017 | |||
| oncogene | ✔ | Budd, W.T. et al., 2015 | |||
| oncogene | ✔ | Poliseno, L. et al., 2010 |
| Tumor Type | Amplification/Deletion | miR-22 | miR-132 | miR-212 |
|---|---|---|---|---|
| All tumors | amp | 97 | 94 | 94 |
| del | 131 | 146 | 146 | |
| ratio | 0.74 | 0.64 | 0.64 | |
| HCC | amp | 2 | 1 | 1 |
| del | 8 | 8 | 8 | |
| ratio | 0.25 | 0.125 | 0.125 | |
| BRCA | amp | 5 | 11 | 11 |
| del | 8 | 20 | 20 | |
| ratio | 0.625 | 0.55 | 0.55 | |
| PC | amp | 8 | 3 | 3 |
| del | 8 | 7 | 7 | |
| ratio | 1 | 0.375 | 0.375 |
| A | B | Neither | A Not B | B Not A | Both | p-Value | Tendency | Fisher Test | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | miR-22 | miR-132 | 41760 | 36 | 36 | 177 | <0.001 | Co-occurrence | p-value > 0.05 | p-value | |
| 2 | miR-22 | miR-212 | 41762 | 36 | 34 | 177 | <0.001 | Co-occurrence | p-value < 0.05 | ||
| 3 | miR-132 | miR-212 | 41796 | 2 | 0 | 211 | <0.001 | Co-occurrence | <0.05 | ||
| 4 | miR22HG | miR-22 | 41769 | 27 | 8 | 205 | <0.001 | Co-occurrence | |||
| 5 | miR22HG | miR-132 | 41761 | 35 | 16 | 197 | <0.001 | Co-occurrence | |||
| 6 | miR22HG | miR-212 | 41763 | 35 | 14 | 197 | <0.001 | Co-occurrence | |||
| 7 | HIC1 | miR-22 | 47611 | 73 | 38 | 190 | <0.001 | Co-occurrence | |||
| 8 | HIC1 | miR-132 | 47648 | 23 | 1 | 240 | <0.001 | Co-occurrence | |||
| 9 | HIC1 | miR-212 | 47648 | 25 | 1 | 238 | <0.001 | Co-occurrence | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Centomo, M.L.; Vitiello, M.; Poliseno, L.; Pandolfi, P.P. An Immunocompetent Environment Unravels the Proto-Oncogenic Role of miR-22. Cancers 2022, 14, 6255. https://doi.org/10.3390/cancers14246255
Centomo ML, Vitiello M, Poliseno L, Pandolfi PP. An Immunocompetent Environment Unravels the Proto-Oncogenic Role of miR-22. Cancers. 2022; 14(24):6255. https://doi.org/10.3390/cancers14246255
Chicago/Turabian StyleCentomo, Maria Laura, Marianna Vitiello, Laura Poliseno, and Pier Paolo Pandolfi. 2022. "An Immunocompetent Environment Unravels the Proto-Oncogenic Role of miR-22" Cancers 14, no. 24: 6255. https://doi.org/10.3390/cancers14246255