
High-Frequency Nanosecond Bleomycin Electrochemotherapy and its Effects on Changes in the Immune System and Survival
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Luciferase Expressing LLC1 Carcinoma Cells
2.2. Mice and Tumour Induction
2.3. Electroporation
2.4. Experimental Scheme
2.5. Evaluation of Tumour Sizes
2.6. Flow Cytometry
2.7. Determination of Specific Antitumour Antibodies
2.8. Statistical Analysis
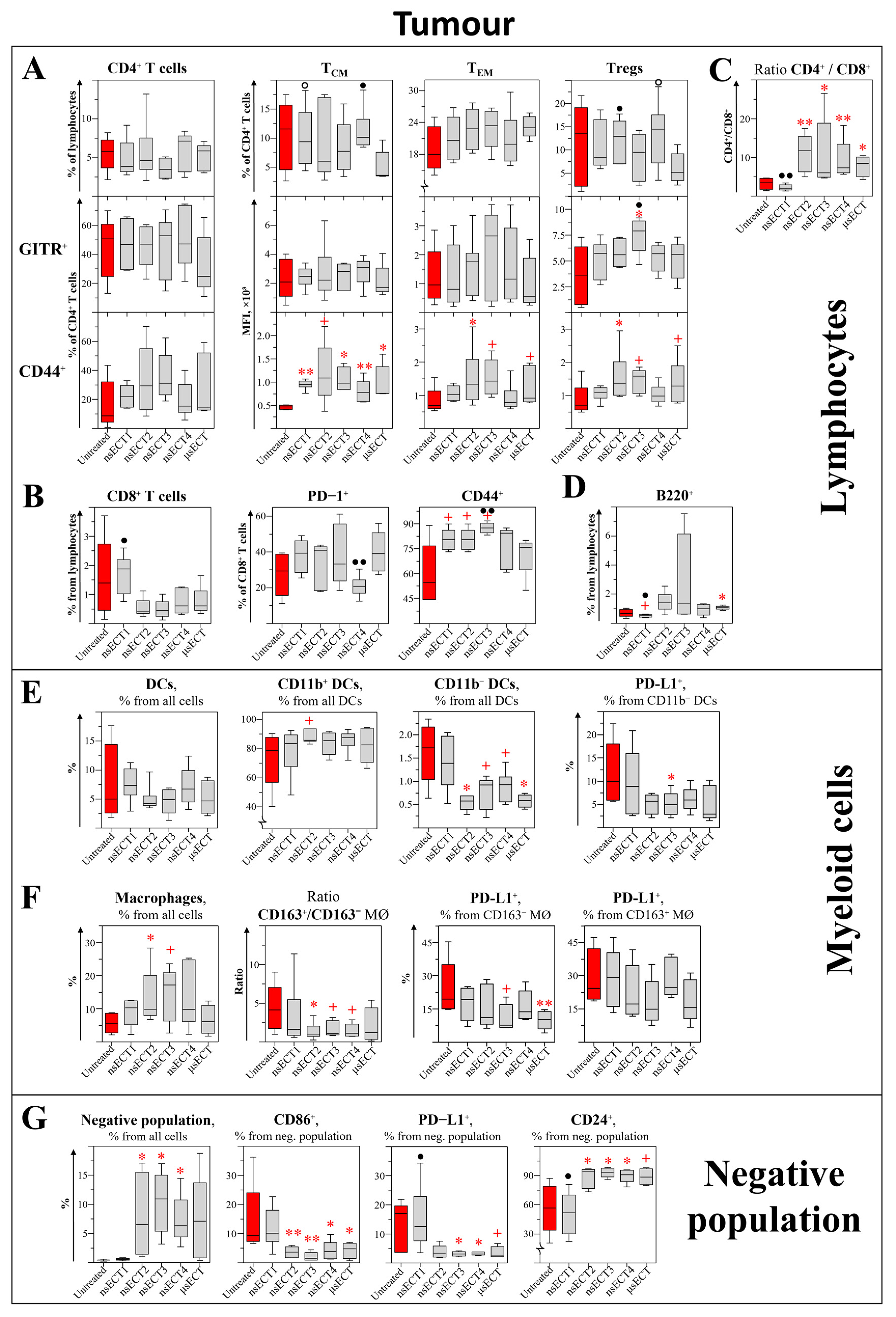
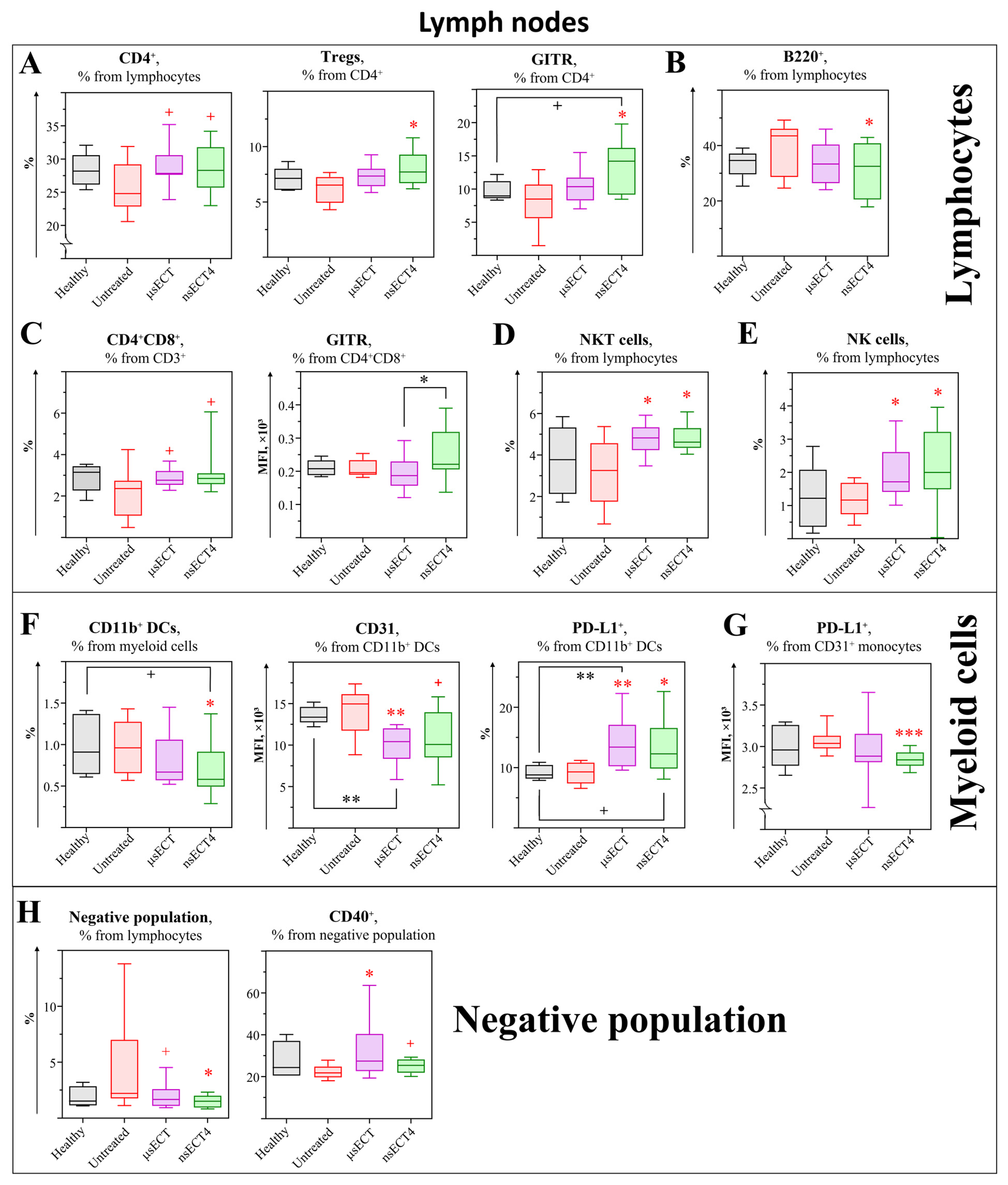
3. Results
3.1. Bleomycin ECT Decreased Tumour Growth and Increased Mice Survival
3.2. Electrochemotherapy with Bleomycin Modulates Cellular Immunity
3.3. Bleomycin nsECT STIMULATES the formation of Humoral Antitumour Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dehez, F.; Delemotte, L.; Kramar, P.; Miklavčič, D.; Tarek, M. Evidence of Conducting Hydrophobic Nanopores across Membranes in Response to an Electric Field. J. Phys. Chem. C 2014, 118, 6752–6757. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.C.; Smith, K.C.; Esser, A.T.; Son, R.S.; Gowrishankar, T.R. A Brief Overview of Electroporation Pulse Strength-Duration Space: A Region Where Additional Intracellular Effects Are Expected. Bioelectrochemistry 2012, 87, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rols, M.P. Gene Delivery by Electroporation in Vitro: Mechanisms. Handb. Electroporation 2017, 1, 387–401. [Google Scholar]
- Shi, J.; Ma, Y.; Zhu, J.; Chen, Y.; Sun, Y.; Yao, Y.; Yang, Z.; Xie, J. A Review on Electroporation-Based Intracellular Delivery. Molecules 2018, 23, 3044. [Google Scholar] [CrossRef] [Green Version]
- Yarmush, M.L.; Golberg, A.; Serša, G.; Kotnik, T.; Miklavčič, D. Electroporation-Based Technologies for Medicine: Principles, Applications, and Challenges. Annu. Rev. Biomed. Eng. 2014, 16, 295–320. [Google Scholar] [CrossRef] [Green Version]
- Gehl, J.; Marty, M.; Sersa, G.; Re, J.; Geertsen, P.F.; Collins, C.G.; Snoj, M.; Larkin, J.O.; Miklavcic, D.; Pavlovic, I.; et al. Electrochemotherapy—An Easy, Highly Effective and Safe Treatment of Cutaneous and Subcutaneous Metastases: Results of ESOPE (European Standard Operating Procedures of Electrochemotherapy) Study. Eur. J. Cancer Suppl. 2006, 4, 3–13. [Google Scholar] [CrossRef]
- Calvet, C.Y.; Mir, L.M. The Promising Alliance of Anti-Cancer Electrochemotherapy with Immunotherapy. Cancer Metastasis Rev. 2016, 35, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Novickij, V.; Baleviciute, A.; Malysko, V.; Zelvys, A.; Radzeviciute, E.; Kos, B.; Zinkeviciene, A.; Miklavcic, D.; Novickij, J.; Girkontaite, I. Effects of Time Delay Between Unipolar Pulses in High Frequency Nano-Electrochemotherapy. IEEE Trans. Biomed. Eng. 2022, 69, 1726–1732. [Google Scholar] [CrossRef]
- Guo, S.; Jing, Y.; Burcus, N.I.; Lassiter, B.P.; Tanaz, R.; Heller, R.; Beebe, S.J. Nano-Pulse Stimulation Induces Potent Immune Responses, Eradicating Local Breast Cancer While Reducing Distant Metastases. Int. J. Cancer 2018, 142, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Chen, X.; Hu, C.; Zhang, X.; Hu, Z.; Yu, J.; Feng, X.; Jiang, K.; Ye, S.; Shen, K.; et al. Nanosecond Pulsed Electric Field (NsPEF) Treatment for Hepatocellular Carcinoma: A Novel Locoregional Ablation Decreasing Lung Metastasis. Cancer Lett. 2014, 346, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Pliquett, U. Joule Heating during Solid Tissue Electroporation. Med. Biol. Eng. Comput. 2003, 41, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Pliquett, U.; Nuccitelli, R. Measurement and Simulation of Joule Heating during Treatment of B-16 Melanoma Tumors in Mice with Nanosecond Pulsed Electric Fields. Bioelectrochemistry 2014, 100, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Xu, J.; Tang, X.; Bian, C.; Liu, H.; Yang, Q.; Tang, J. Scaling Relationship of In Vivo Muscle Contraction Strength of Rabbits Exposed to High-Frequency Nanosecond Pulse Bursts. Technol. Cancer Res. Treat. 2018, 17, 8078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizintin, A.; Markovic, S.; Scancar, J.; Miklavcic, D. Electroporation with Nanosecond Pulses and Bleomycin or Cisplatin Results in Efficient Cell Kill and Low Metal Release from Electrodes. Bioelectrochemistry 2021, 140, 107798. [Google Scholar] [CrossRef] [PubMed]
- Simsek, C.; Esin, E.; Yalcin, S. Metronomic Chemotherapy: A Systematic Review of the Literature and Clinical Experience. J. Oncol. 2019, 2019, 5483791. [Google Scholar] [CrossRef] [Green Version]
- Koumarianou, A.; Christodoulou, M.-I.; Patapis, P.; Papadopoulos, I.; Liakata, E.; Giagini, A.; Stavropoulou, A.; Poulakaki, N.; Tountas, N.; Xiros, N.; et al. The Effect of Metronomic versus Standard Chemotherapy on the Regulatory to Effector T-Cell Equilibrium in Cancer Patients. Exp. Hematol. Oncol. 2014, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic Capecitabine as an Immune Modulator in Glioblastoma Patients Reduces Myeloid-Derived Suppressor Cells. JCI Insight 2019, 4, e130748. [Google Scholar] [CrossRef] [Green Version]
- Radzevičiūtė, E.; Malyško-Ptašinskė, V.; Kulbacka, J.; Rembiałkowska, N.; Novickij, J.; Girkontaitė, I.; Novickij, V. Nanosecond Electrochemotherapy Using Bleomycin or Doxorubicin: Influence of Pulse Amplitude, Duration and Burst Frequency. Bioelectrochemistry 2022, 148, 108251. [Google Scholar] [CrossRef]
- Novickij, V.; Malyško, V.; Želvys, A.; Balevičiūtė, A.; Zinkevičienė, A.; Novickij, J.; Girkontaitė, I.; Balevičiūte, A.; Zinkevičiene, A.; Novickij, J.; et al. Electrochemotherapy Using Doxorubicin and Nanosecond Electric Field Pulses: A Pilot in Vivo Study. Molecules 2020, 25, 4601. [Google Scholar] [CrossRef]
- Vizintin, A.; Markovic, S.; Scancar, J.; Kladnik, J.; Turel, I.; Miklavcic, D. Nanosecond Electric Pulses Are Equally Effective in Electrochemotherapy with Cisplatin as Microsecond Pulses. Radiol. Oncol. 2022, 56, 326–335. [Google Scholar] [CrossRef]
- Mir, L.M.; Belehradek, M.; Domenge, C.; Orlowski, S.; Poddevin, B.; Belehradek, J.J.; Schwaab, G.; Luboinski, B.; Paoletti, C. Electrochemotherapy, a new antitumor treatment: First clinical trial. Comptes Rendus L’academie Des Sci. Ser. III Sci. 1991, 313, 613–618. [Google Scholar]
- Sersa, G.; Miklavcic, D.; Cemazar, M.; Rudolf, Z.; Pucihar, G.; Snoj, M. Electrochemotherapy in Treatment of Tumours. Eur. J. Surg. Oncol. EJSO 2008, 34, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Snoj, M.; Cemazar, M.; Srnovrsnik, T.; Kosir, S.P.; Sersa, G. Limb Sparing Treatment of Bleeding Melanoma Recurrence by Electrochemotherapy. Tumori J. 2009, 95, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Kranjc, S.; Cemazar, M.; Grosel, A.; Sentjurc, M.; Sersa, G. Radiosensitising Effect of Electrochemotherapy with Bleomycin in LPB Sarcoma Cells and Tumors in Mice. BMC Cancer 2005, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Gothelf, A.; Mir, L.M.; Gehl, J. Electrochemotherapy: Results of Cancer Treatment Using Enhanced Delivery of Bleomycin by Electroporation. Cancer Treat. Rev. 2003, 29, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stubbe, J. Bleomycins: Towards Better Therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin Pathways: Pharmacodynamics and Adverse Effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Jaroszeski, M.J.; Dang, V.; Pottinger, C.; Hickey, J.; Gilbert, R.; Heller, R. Toxicity of Anticancer Agents Mediated by Electroporation in Vitro. Anticancer Drugs 2000, 11, 201–208. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Geboers, B.; Scheffer, H.J.; Graybill, P.M.; Ruarus, A.H.; Nieuwenhuizen, S.; Puijk, R.S.; van den Tol, P.M.; Davalos, R.V.; Rubinsky, B.; de Gruijl, T.D.; et al. High-Voltage Electrical Pulses in Oncology: Irreversible Electroporation, Electrochemotherapy, Gene Electrotransfer, Electrofusion, and Electroimmunotherapy. Radiology 2020, 295, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, J.S.S.; Ray, P.; Hayashi, T.; Whisenant, T.C.; Vicente, D.; Carson, D.A.; Miller, A.M.; Schoenberger, S.P.; White, R.R. Irreversible Electroporation Combined with Checkpoint Blockade and TLR7 Stimulation Induces Antitumor Immunity in a Murine Pancreatic Cancer Model. Cancer Immunol. Res. 2019, 7, 1714–1726. [Google Scholar] [CrossRef]
- Yang, J.; Eresen, A.; Shangguan, J.; Ma, Q.; Yaghmai, V.; Zhang, Z. Irreversible Electroporation Ablation Overcomes Tumor-Associated Immunosuppression to Improve the Efficacy of DC Vaccination in a Mice Model of Pancreatic Cancer. Oncoimmunology 2021, 10, 1875638. [Google Scholar] [CrossRef] [PubMed]
- Novickij, V.; Česna, R.; Perminaite, E.; Zinkevičiene, A.; Characiejus, D.; Novickij, J.; Šatkauskas, S.; Ruzgys, P.; Girkontaite, I. Antitumor Response and Immunomodulatory Effects of Sub-Microsecond Irreversible Electroporation and Its Combination with Calcium Electroporation. Cancers 2019, 11, 1763. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Sun, S.; Zhang, Y.; Xie, F.; Li, S. The Role of Irreversible Electroporation in Promoting M1 Macrophage Polarization via Regulating the HMGB1-RAGE-MAPK Axis in Pancreatic Cancer. Oncoimmunology 2021, 10, 1897295. [Google Scholar] [CrossRef]
- He, C.; Huang, X.; Zhang, Y.; Lin, X.; Li, S. T-cell Activation and Immune Memory Enhancement Induced by Irreversible Electroporation in Pancreatic Cancer. Clin. Transl. Med. 2020, 10, e39. [Google Scholar] [CrossRef]
- Calvet, C.Y.; Famin, D.; André, F.M.; Mir, L.M. Electrochemotherapy with Bleomycin Induces Hallmarks of Immunogenic Cell Death in Murine Colon Cancer Cells. Oncoimmunology 2014, 3, e28131. [Google Scholar] [CrossRef] [Green Version]
- Torrero, M.N.; Henk, W.G.; Li, S. Regression of High-Grade Malignancy in Mice by Bleomycin and Interleukin-12 Electrochemogenetherapy. Clin. Cancer Res. 2006, 12, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Markelc, B.; Sersa, G.; Cemazar, M. Differential Mechanisms Associated with Vascular Disrupting Action of Electrochemotherapy: Intravital Microscopy on the Level of Single Normal and Tumor Blood Vessels. PLoS ONE 2013, 8, e59557. [Google Scholar] [CrossRef] [Green Version]
- Mekid, H.; Tounekti, O.; Spatz, A.; Cemazar, M.; el Kebir, F.Z.; Mir, L.M. In Vivo Evolution of Tumour Cells after the Generation of Double-Strand DNA Breaks. Br. J. Cancer 2003, 88, 1763–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Bernat, C.; Al-Sakere, B.; Ghiringhelli, F.; Opolon, P.; Carpentier, A.F.; Zitvogel, L.; Mir, L.M.; Robert, C. Tumor Destruction Using Electrochemotherapy Followed by CpG Oligodeoxynucleotide Injection Induces Distant Tumor Responses. Cancer Immunol. Immunother. 2008, 57, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Tremble, L.F.; O’Brien, M.A.; Soden, D.M.; Forde, P.F. Electrochemotherapy with Cisplatin Increases Survival and Induces Immunogenic Responses in Murine Models of Lung Cancer and Colorectal Cancer. Cancer Lett. 2019, 442, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Ursic, K.; Kos, S.; Kamensek, U.; Cemazar, M.; Scancar, J.; Bucek, S.; Kranjc, S.; Staresinic, B.; Sersa, G. Comparable Effectiveness and Immunomodulatory Actions of Oxaliplatin and Cisplatin in Electrochemotherapy of Murine Melanoma. Bioelectrochemistry 2018, 119, 161–171. [Google Scholar] [CrossRef]
- Serša, G.; Miklavčič, D.; Čemažar, M.; Belehradek, J.; Jarm, T.; Mir, L.M. Electrochemotherapy with CDDP on LPB Sarcoma: Comparison of the Anti- Tumor Effectiveness in Immunocompetent and Immunodeficient Mice. Bioelectrochem. Bioenerg. 1997, 43, 279–283. [Google Scholar] [CrossRef]
- Chen, R.; Sain, N.M.; Harlow, K.T.; Chen, Y.-J.; Shires, P.K.; Heller, R.; Beebe, S.J. A Protective Effect after Clearance of Orthotopic Rat Hepatocellular Carcinoma by Nanosecond Pulsed Electric Fields. Eur. J. Cancer 2014, 50, 2705–2713. [Google Scholar] [CrossRef]
- Nuccitelli, R.; Berridge, J.C.; Mallon, Z.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanoelectroablation of Murine Tumors Triggers a CD8-Dependent Inhibition of Secondary Tumor Growth. PLoS ONE 2015, 10, e0134364. [Google Scholar] [CrossRef] [Green Version]
- Skeate, J.G.; da Silva, D.M.; Chavez-Juan, E.; Anand, S.; Nuccitelli, R.; Martin Kast, W. Nano-Pulse Stimulation Induces Immunogenic Cell Death in Human Papillomavirus-Transformed Tumors and Initiates an Adaptive Immune Response. PLoS ONE 2018, 13, e0191311. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R.; Tran, K.; Lui, K.; Huynh, J.; Athos, B.; Kreis, M.; Nuccitelli, P.; de Fabo, E.C. Non-Thermal Nanoelectroablation of UV-Induced Murine Melanomas Stimulates an Immune Response. Pigment Cell Melanoma Res. 2012, 25, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yin, S.; Hu, C.; Chen, X.; Jiang, K.; Ye, S.; Feng, X.; Fan, S.; Xie, H.; Zhou, L.; et al. Comparative Study of Nanosecond Electric Fields in Vitro and in Vivo on Hepatocellular Carcinoma Indicate Macrophage Infiltration Contribute to Tumor Ablation in Vivo. PLoS ONE 2014, 9, e0086421. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, S.; Zhu, L.; Zhang, L.; Liu, J.; Xu, D.; Tian, G.; Jiang, T. Antitumor Effect and Immune Response of Nanosecond Pulsed Electric Fields in Pancreatic Cancer. Front. Oncol. 2021, 10, 621092. [Google Scholar] [CrossRef] [PubMed]
- Ringel-Scaia, V.M.; Beitel-White, N.; Lorenzo, M.F.; Brock, R.M.; Huie, K.E.; Coutermarsh-Ott, S.; Eden, K.; McDaniel, D.K.; Verbridge, S.S.; Rossmeisl, J.H.; et al. High-Frequency Irreversible Electroporation Is an Effective Tumor Ablation Strategy That Induces Immunologic Cell Death and Promotes Systemic Anti-Tumor Immunity. eBioMedicine 2019, 44, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Novickij, V.; Grainys, A.; Butkus, P.; Tolvaišienė, S.; Švedienė, J.; Paškevičius, A.; Novickij, J. High-Frequency Submicrosecond Electroporator. Biotechnol. Biotechnol. Equip. 2016, 30, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Cemazar, M.; Sersa, G. Recent Advances in Electrochemotherapy. Bioelectricity 2019, 1, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Matthiessen, L.W.; Chalmers, R.L.; Sainsbury, D.C.G.; Veeramani, S.; Kessell, G.; Humphreys, A.C.; Bond, J.E.; Muir, T.; Gehl, J. Management of Cutaneous Metastases Using Electrochemotherapy. Acta Oncol. 2011, 50, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Plaschke, C.C.; Bertino, G.; McCaul, J.A.; Grau, J.J.; de Bree, R.; Sersa, G.; Occhini, A.; Groselj, A.; Langdon, C.; Heuveling, D.A.; et al. European Research on Electrochemotherapy in Head and Neck Cancer (EURECA) Project: Results from the Treatment of Mucosal Cancers. Eur. J. Cancer 2017, 87, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Bardet, S.M.; Carr, L.; Soueid, M.; Arnaud-Cormos, D.; Leveque, P.; O’Connor, R.P. Multiphoton Imaging Reveals That Nanosecond Pulsed Electric Fields Collapse Tumor and Normal Vascular Perfusion in Human Glioblastoma Xenografts. Sci. Rep. 2016, 6, 34443. [Google Scholar] [CrossRef]
- Mi, Y.; Rui, S.; Li, C.; Yao, C.; Xu, J.; Bian, C.; Tang, X. Multi-Parametric Study of Temperature and Thermal Damage of Tumor Exposed to High-Frequency Nanosecond-Pulsed Electric Fields Based on Finite Element Simulation. Med. Biol. Eng. Comput. 2017, 55, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Miklavcic, D.; Pucihar, G.; Pavlovec, M.; Ribaric, S.; Mali, M.; Macek-Lebar, A.; Petkovsek, M.; Nastran, J.; Kranjc, S.; Cemazar, M.; et al. The Effect of High Frequency Electric Pulses on Muscle Contractions and Antitumor Efficiency in Vivo for a Potential Use in Clinical Electrochemotherapy. Bioelectrochemistry 2005, 65, 121–128. [Google Scholar] [CrossRef]
- Fang, J.J.; Zhu, Z.Y.; Dong, H.; Zheng, G.Q.; Teng, A.G.; Liu, A.J. Effect of Spleen Lymphocytes on the Splenomegaly in Hepatocellular Carcinoma-Bearing Mice. Biomed. Environ. Sci. 2014, 27, 17–26. [Google Scholar] [CrossRef]
- Mackay, W.D. Role of Splenomegaly in Tumour-Bearing Mice. Nature 1965, 205, 918–919. [Google Scholar] [CrossRef]
- duPre’, S.A.; Hunter, K.W. Murine Mammary Carcinoma 4T1 Induces a Leukemoid Reaction with Splenomegaly: Association with Tumor-Derived Growth Factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar] [CrossRef]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 Blockade and GITR Triggering Induce a Potent Antitumor Immunity in Murine Cancer Models and Synergizes with Chemotherapeutic Drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- van Olffen, R.W.; Koning, N.; van Gisbergen, K.P.J.M.; Wensveen, F.M.; Hoek, R.M.; Boon, L.; Hamann, J.; van Lier, R.A.W.; Nolte, M.A. GITR Triggering Induces Expansion of Both Effector and Regulatory CD4 + T Cells In Vivo. J. Immunol. 2009, 182, 7490–7500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchetti, S.; Zollo, O.; Bruscoli, S.; Agostini, M.; Bianchini, R.; Nocentini, G.; Ayroldi, E.; Riccardi, C. Frontline: GITR, a Member of the TNF Receptor Superfamily, Is Costimulatory to Mouse T Lymphocyte Subpopulations. Eur. J. Immunol. 2004, 34, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Nocentini, G.; Riccardi, C.; Pandolfi, P.P. Role of GITR in Activation Response of T Lymphocytes. Blood 2002, 100, 350–352. [Google Scholar] [CrossRef] [Green Version]
- Tabakov, D.V.; Zabotina, T.N.; Chanturia, N.V.; Zakharova, E.N.; Vorotnikov, I.K.; Selchuk, V.Y.; Sokolovskiy, V.V.; Petrovsky, A.V. The Relationship of GITR, Lag-3 and PD-1 Expression with the Main Indicators of Systemic and Local Immunity in Patients with Breast Cancer. J. Mod. Oncol. 2021, 23, 457–465. [Google Scholar] [CrossRef]
- Sales de Sá, R.; Miranda Galvis, M.; Mariz, B.A.L.A.; Leite, A.A.; Schultz, L.; Almeida, O.P.; Santos-Silva, A.R.; Pinto, C.A.L.; Vargas, P.A.; Gollob, K.J.; et al. Increased Tumor Immune Microenvironment CD3+ and CD20+ Lymphocytes Predict a Better Prognosis in Oral Tongue Squamous Cell Carcinoma. Front. Cell. Dev. Biol. 2021, 8, 622161. [Google Scholar] [CrossRef]
- Larsen, S.K.; Gao, Y.; Basse, P.H. NK Cells in the Tumor Microenvironment. Crit. Rev. Oncog. 2014, 19, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, J.H.W.; Koch, J.; Gotz, J.J.; Arnold, A.; Reusch, U.; Gantke, T.; Rajkovic, E.; Treder, M.; Cerwenka, A. Cd16a Activation of Nk Cells Promotes Nk Cell Proliferation and Memory-like Cytotoxicity against Cancer Cells. Cancer Immunol. Res. 2018, 6, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, N.D.; Nutt, S.L.; Carotta, S. Regulation of Murine Natural Killer Cell Commitment. Front. Immunol. 2013, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Crinier, A.; Milpied, P.; Escalière, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986.e5. [Google Scholar] [CrossRef] [PubMed]
- Crinier, A.; Narni-Mancinelli, E.; Ugolini, S.; Vivier, E. SnapShot: Natural Killer Cells. Cell 2020, 180, 1280–1280.e1. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.C.; Fischer, P.; Kakrecha, B.; Linsley, P.S.; Wambre, E.; Liu, M.C.; Rust, B.J.; Lee, D.; Penhallow, B.; Orduno, N.M.; et al. Renal Cell Carcinoma (RCC) Tumors Display Large Expansion of Double Positive (DP) CD4+CD8+ T Cells with Expression of Exhaustion Markers. Front. Immunol. 2018, 9, 2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrot, T.; Oger, R.; Allard, M.; Desfrançois, J.; Raingeard de la Blétière, D.; Coutolleau, A.; Preisser, L.; Khammari, A.; Dréno, B.; Delneste, Y.; et al. Transcriptomic Features of Tumour-Infiltrating CD4lowCD8high Double Positive Aβ T Cells in Melanoma. Sci. Rep. 2020, 10, 5900. [Google Scholar] [CrossRef] [Green Version]
- Pahar, B.; Lackner, A.A.; Veazey, R.S. Intestinal Double-Positive CD4+CD8+ T Cells Are Highly Activated Memory Cells with an Increased Capacity to Produce Cytokines. Eur. J. Immunol. 2006, 36, 583–592. [Google Scholar] [CrossRef]
- van Kaer, L.; Rabacal, W.A.S.; Algood, H.M.S.; Parekh, V.V.; Olivares-Villagómez, D. In Vitro Induction of Regulatory CD4+CD8α+ T Cells by TGF-β, IL-7 and IFN-γ. PLoS ONE 2013, 8, e67821. [Google Scholar] [CrossRef]
- Yeh, S.; Li, Z.; Sen, H.N.; Lim, W.K.; Gill, F.; Perkins, K.; Rao, V.K.; Nussenblatt, R.B. Scleritis and Multiple Systemic Autoimmune Manifestations in Chronic Natural Killer Cell Lymphocytosis Associated with Elevated TCRα/Β+CD3+CD4-CD8- Double-Negative T Cells. Br. J. Ophthalmol. 2010, 94, 748–752. [Google Scholar] [CrossRef]
- Brandt, D.; Sergon, M.; Abraham, S.; Mäbert, K.; Hedrich, C.M. TCR(+)CD3(+)CD4(-)CD8(-) Effector T Cells in Psoriasis. Clin. Immunol. 2017, 181, 51–59. [Google Scholar] [CrossRef]
- Soklic, T.K.; Silar, M.; Rijavec, M.; Koren, A.; Kern, I.; Hocevar-Boltezar, I.; Korosec, P. CD3(+)CD4(-)CD8(-) Mucosal T Cells Are Associated with Uncontrolled Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. 2019, 143, 1235–1237.e5. [Google Scholar] [CrossRef] [Green Version]
- di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; de Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Koike, T.; Harada, K.; Horiuchi, S.; Kitamura, D. The Quantity of CD40 Signaling Determines the Differentiation of B Cells into Functionally Distinct Memory Cell Subsets. Elife 2019, 8, e44245. [Google Scholar] [CrossRef] [PubMed]
- Munroe, M.E.; Bishop, G.A. A Costimulatory Function for T Cell CD40. J. Immunol. 2007, 178, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimabukuro-Vornhagen, A.; Schlößer, H.A.; Gryschok, L.; Malcher, J.; Wennhold, K.; Garcia-Marquez, M.; Herbold, T.; Neuhaus, L.S.; Becker, H.J.; Fiedler, A.; et al. Characterization of Tumor-Associated B-Cell Subsets in Patients with Colorectal Cancer. Oncotarget 2014, 5, 4651–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, S.; Candido, J.B.; McDermott, J.R.; Ghirelli, C.; Maniati, E.; Beers, S.A.; Balkwill, F.R.; Kocher, H.M.; Capasso, M. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Front. Immunol. 2019, 10, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Q.; O’Flanagan, S.; Lam, T.; Roy, P.; Pelaez, F.; Burbach, B.J.; Azarin, S.M.; Shimizu, Y.; Bischof, J.C. Engineering T Cell Response to Cancer Antigens by Choice of Focal Therapeutic Conditions. Int. J. Hyperth. 2019, 36, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Alinezhadbalalami, N.; Graybill, P.M.; Imran, K.M.; Verbridge, S.S.; Allen, I.C.; Davalos, R. v Generation of Tumor-Activated T Cells Using Electroporation. Bioelectrochemistry 2021, 142, 107886. [Google Scholar] [CrossRef]
- Laine, P.; Meurman, J.H.; Tenovuo, J.; Murtomaa, H.; Lindqvist, C.; Pyrhönen, S.; Teerenhovi, L. Salivary Flow and Composition in Lymphoma Patients before, during and after Treatment with Cytostatic Drugs. Eur. J. Cancer B Oral Oncol. 1992, 28, 125–128. [Google Scholar] [CrossRef]
- Ito, K.; Okamoto, M.; Inaguma, Y.; Okamoto, A.; Ando, M.; Ando, Y.; Tsuge, M.; Tomono, A.; Kakumae, Y.; Hayashi, T.; et al. Influence of R-CHOP Therapy on Immune System Restoration in Patients with B-Cell Lymphoma. Oncology 2016, 91, 302–310. [Google Scholar] [CrossRef]
- Halliley, J.L.; Tipton, C.M.; Liesveld, J.; Rosenberg, A.F.; Darce, J.; Gregoretti, I.V.; Popova, L.; Kaminiski, D.; Fucile, C.F.; Albizua, I.; et al. Long-Lived Plasma Cells Are Contained within the CD19(-)CD38(Hi)CD138(+) Subset in Human Bone Marrow. Immunity 2015, 43, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Good-Jacobson, K.L.; Szumilas, C.G.; Chen, L.; Sharpe, A.H.; Tomayko, M.M.; Shlomchik, M.J. PD-1 Regulates Germinal Center B Cell Survival and the Formation and Affinity of Long-Lived Plasma Cells. Nat. Immunol. 2010, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Fornasa, G.; Guedj, K.; ben Mkaddem, S.; Gaston, A.-T.; Khallou-Laschet, J.; Morvan, M.; Nicoletti, A.; Caligiuri, G. CD31 Is a Key Coinhibitory Receptor in the Development of Immunogenic Dendritic Cells. Proc. Natl. Acad. Sci. USA 2014, 111, E1101–E1110. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef] [PubMed]
- Goudin, N.; Chappert, P.; Mégret, J.; Gross, D.-A.; Rocha, B.; Azogui, O. Depletion of Regulatory T Cells Induces High Numbers of Dendritic Cells and Unmasks a Subset of Anti-Tumour CD8+CD11c+ PD-1lo Effector T Cells. PLoS ONE 2016, 11, e0157822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, J.K.; Gowthaman, U.; Zhang, B.; Mattsson, J.; Szeponik, L.; Liu, D.; Wu, R.; White, T.; Calabro, S.; Xu, L.; et al. Migratory CD11b+ Conventional Dendritic Cells Induce T Follicular Helper Cell–Dependent Antibody Responses. Sci. Immunol. 2017, 2, 9169. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Edwards, C.M.; Mundy, G.R. Gr-1+CD11b+ Myeloid-Derived Suppressor Cells: Formidable Partners in Tumor Metastasis. J. Bone Miner. Res. 2010, 25, 1701–1706. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Chu, T.H.; Nienhüser, H.; Jiang, Z.; del Portillo, A.; Remotti, H.E.; White, R.A.; Hayakawa, Y.; Tomita, H.; Fox, J.G.; et al. PD-1 Signaling Promotes Tumor-Infiltrating Myeloid-Derived Suppressor Cells and Gastric Tumorigenesis in Mice. Gastroenterology 2021, 160, 781–796. [Google Scholar] [CrossRef]
- Wang, Y.; Schafer, C.C.; Hough, K.P.; Tousif, S.; Duncan, S.R.; Kearney, J.F.; Ponnazhagan, S.; Hsu, H.-C.; Deshane, J.S. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J. Immunol. 2018, 201, 278–295. [Google Scholar] [CrossRef] [Green Version]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 Macrophages in Tumor Islets and Stroma in Relation to Prognosis of Non-Small Cell Lung Cancer. BMC Immunol 2018, 19, 3. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, J.S.; Papadopoulos, J.; Seung, W.K.; Maya, M.; Zhang, F.; He, J.; Fan, D.; Langley, R.; Fidler, I.J. Circulating Monocytes Expressing CD31: Implications for Acute and Chronic Angiogenesis. Am. J. Pathol. 2009, 174, 1972–1980. [Google Scholar] [CrossRef] [Green Version]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like Macrophages Change Tumor Blood Vessels and Microenvironment in Murine Melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef] [PubMed]
- Merchand-Reyes, G.; Robledo-Avila, F.H.; Buteyn, N.J.; Gautam, S.; Santhanam, R.; Fatehchand, K.; Mo, X.; Partida-Sanchez, S.; Butchar, J.P.; Tridandapani, S. CD31 Acts as a Checkpoint Molecule and Is Modulated by FcγR-Mediated Signaling in Monocytes. J. Immunol. 2019, 203, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, K.; Moriyama, M.; Furukawa, S.; Rafiul, H.A.S.M.; Maruse, Y.; Jinno, T.; Tanaka, A.; Ohta, M.; Ishiguro, N.; Yamauchi, M.; et al. CD163+CD204+ Tumor-Associated Macrophages Contribute to T Cell Regulation via Interleukin-10 and PD-L1 Production in Oral Squamous Cell Carcinoma. Sci. Rep. 2017, 7, 1755. [Google Scholar] [CrossRef] [Green Version]
- Fischer-Riepe, L.; Daber, N.; Schulte-Schrepping, J.; Véras De Carvalho, B.C.; Russo, A.; Pohlen, M.; Fischer, J.; Chasan, A.I.; Wolf, M.; Ulas, T.; et al. CD163 Expression Defines Specific, IRF8-Dependent, Immune-Modulatory Macrophages in the Bone Marrow. J. Allergy Clin. Immunol. 2020, 146, 1137–1151. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Choi, K.U.; Kim, A.; Lee, S.J.; Lee, J.H.; Suh, D.S.; Kwon, B.S.; Hwang, C. PD-L1 Expression on Stromal Tumor-Infiltrating Lymphocytes Is a Favorable Prognostic Factor in Ovarian Serous Carcinoma. J. Ovarian Res. 2019, 12, 56. [Google Scholar] [CrossRef]
- Ferrata, M.; Schad, A.; Zimmer, S.; Musholt, T.J.; Bahr, K.; Kuenzel, J.; Becker, S.; Springer, E.; Roth, W.; Weber, M.M.; et al. PD-L1 Expression and Immune Cell Infiltration in Gastroenteropancreatic (GEP) and Non-GEP Neuroendocrine Neoplasms with High Proliferative Activity. Front. Oncol. 2019, 9, 343. [Google Scholar] [CrossRef] [Green Version]
- Gool, S.W.; Vandenberghe, P.; de Boer, M.; Ceuppens, J.L. CD80, CD86 and CD40 Provide Accessory Signals in a Multiple-Step T-Cell Activation Model. Immunol. Rev. 1996, 153, 47–83. [Google Scholar] [CrossRef]
- Damoiseaux, J.G.M.C.; Yagita, H.; Okumura, K.; van Breda Vriesman, P.J.C. Costimulatory Molecules CD80 and CD86 in the Rat; Tissue Distribution and Expression by Antigen-Presenting Cells. J. Leukoc. Biol. 1998, 64, 803–809. [Google Scholar] [CrossRef]
- Tang, M.-R.; Guo, J.-Y.; Wang, D.; Xu, N. Identification of CD24 as a Marker for Tumorigenesis of Melanoma. Onco-Targets Ther. 2018, 11, 3401–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, M.; Chang, A. Ligation of CD24 on T Cells and B Cells Modulates the Cytokine Production Profile of Activated Tumor-Draining Lymph Node Cells. Cancer Res. 2008, 68, 4627. [Google Scholar]
- Olesch, C.; Brunn, D.; Aktay-Cetin, Ö.; Sirait-Fischer, E.; Pullamsetti, S.S.; Grimminger, F.; Seeger, W.; Brüne, B.; Weigert, A.; Savai, R. Picturing of the Lung Tumor Cellular Composition by Multispectral Flow Cytometry. Front. Immunol. 2022, 13, 827719. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | Pulse Parameters | Energy (J) |
|---|---|---|
| nsPEF1 | 3.5 kV/cm × 200 ns × 200, 1 kHz | ~0.5 |
| nsPEF2 | 3.5 kV/cm × 200 ns × 200, 1 MHz | ~0.5 |
| nsPEF3 | 3.5 kV/cm × 700 ns × 200, 1 kHz | ~1.6 |
| nsPEF4 | 3.5 kV/cm × 700 ns × 200, 1 MHz | ~1.6 |
| μsPEF | 1.3 kV/cm × 100 μs × 8, 1 Hz | ~0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balevičiūtė, A.; Radzevičiūtė, E.; Želvys, A.; Malyško-Ptašinskė, V.; Novickij, J.; Zinkevičienė, A.; Kašėta, V.; Novickij, V.; Girkontaitė, I. High-Frequency Nanosecond Bleomycin Electrochemotherapy and its Effects on Changes in the Immune System and Survival. Cancers 2022, 14, 6254. https://doi.org/10.3390/cancers14246254
Balevičiūtė A, Radzevičiūtė E, Želvys A, Malyško-Ptašinskė V, Novickij J, Zinkevičienė A, Kašėta V, Novickij V, Girkontaitė I. High-Frequency Nanosecond Bleomycin Electrochemotherapy and its Effects on Changes in the Immune System and Survival. Cancers. 2022; 14(24):6254. https://doi.org/10.3390/cancers14246254
Chicago/Turabian StyleBalevičiūtė, Austėja, Eivina Radzevičiūtė, Augustinas Želvys, Veronika Malyško-Ptašinskė, Jurij Novickij, Auksė Zinkevičienė, Vytautas Kašėta, Vitalij Novickij, and Irutė Girkontaitė. 2022. "High-Frequency Nanosecond Bleomycin Electrochemotherapy and its Effects on Changes in the Immune System and Survival" Cancers 14, no. 24: 6254. https://doi.org/10.3390/cancers14246254