
Drug Inhibition of Redox Factor-1 Restores Hypoxia-Driven Changes in Tuberous Sclerosis Complex 2 Deficient Cells
 , and
, and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Biochemicals
2.2. Cell Culture, Transfection, and Preparing Cell Lysates
2.3. Western Blotting
2.4. Anchorage-Independent Cell Growth in Soft Agar
2.5. Vasculature Mimicry
2.6. Cytotoxicity Assays: Assays for Spheroids, Outgrowth, and Acridine Orange/Propidium Iodide (AO/PI) Cell Viability
2.7. Transcription Assays
2.8. Targeted Metabolic Profiling
2.9. Oxygen Consumption and Extracellular Acidification Rate
2.10. Experimental Repeats and Statistical Analysis
3. Results
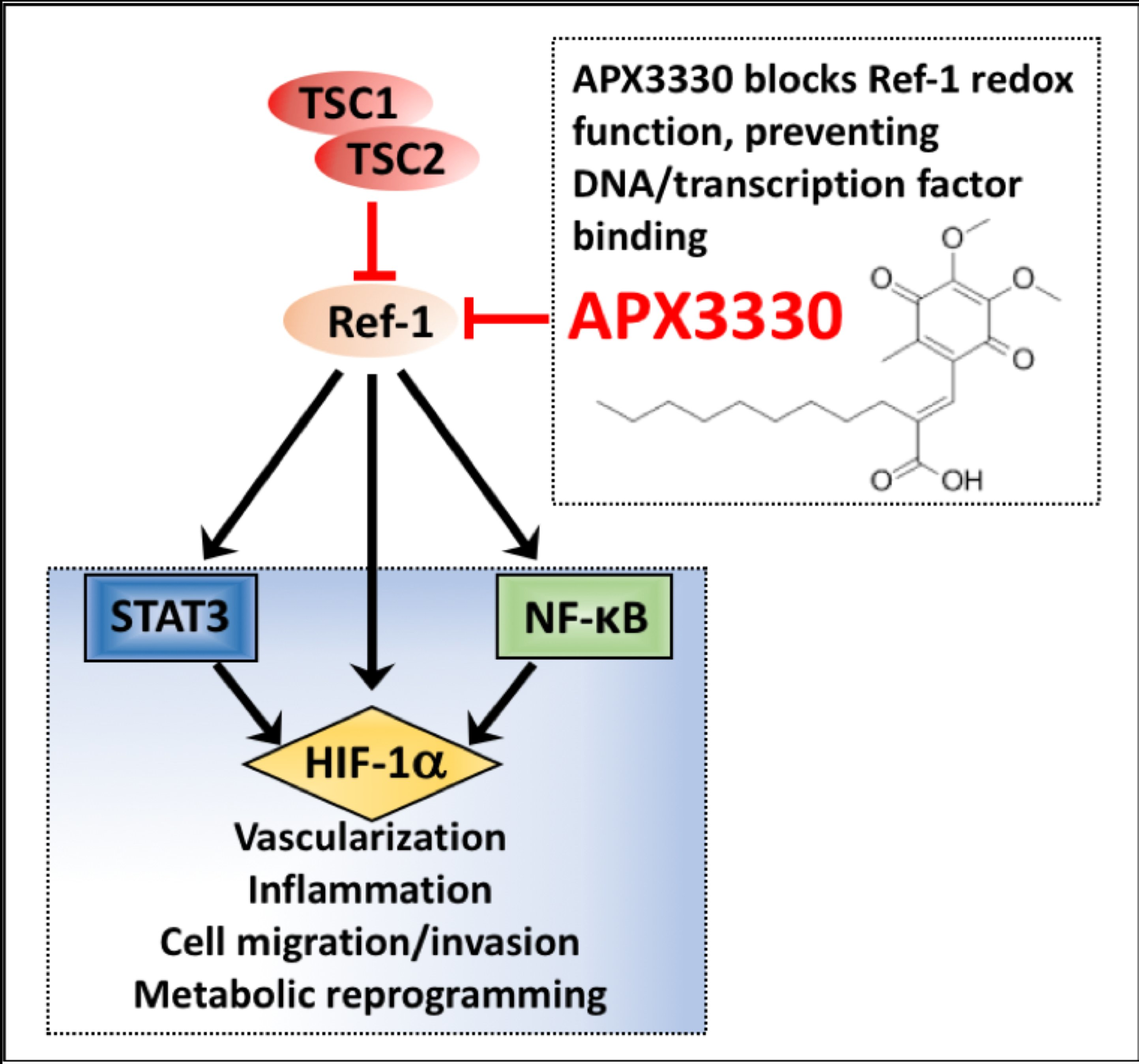
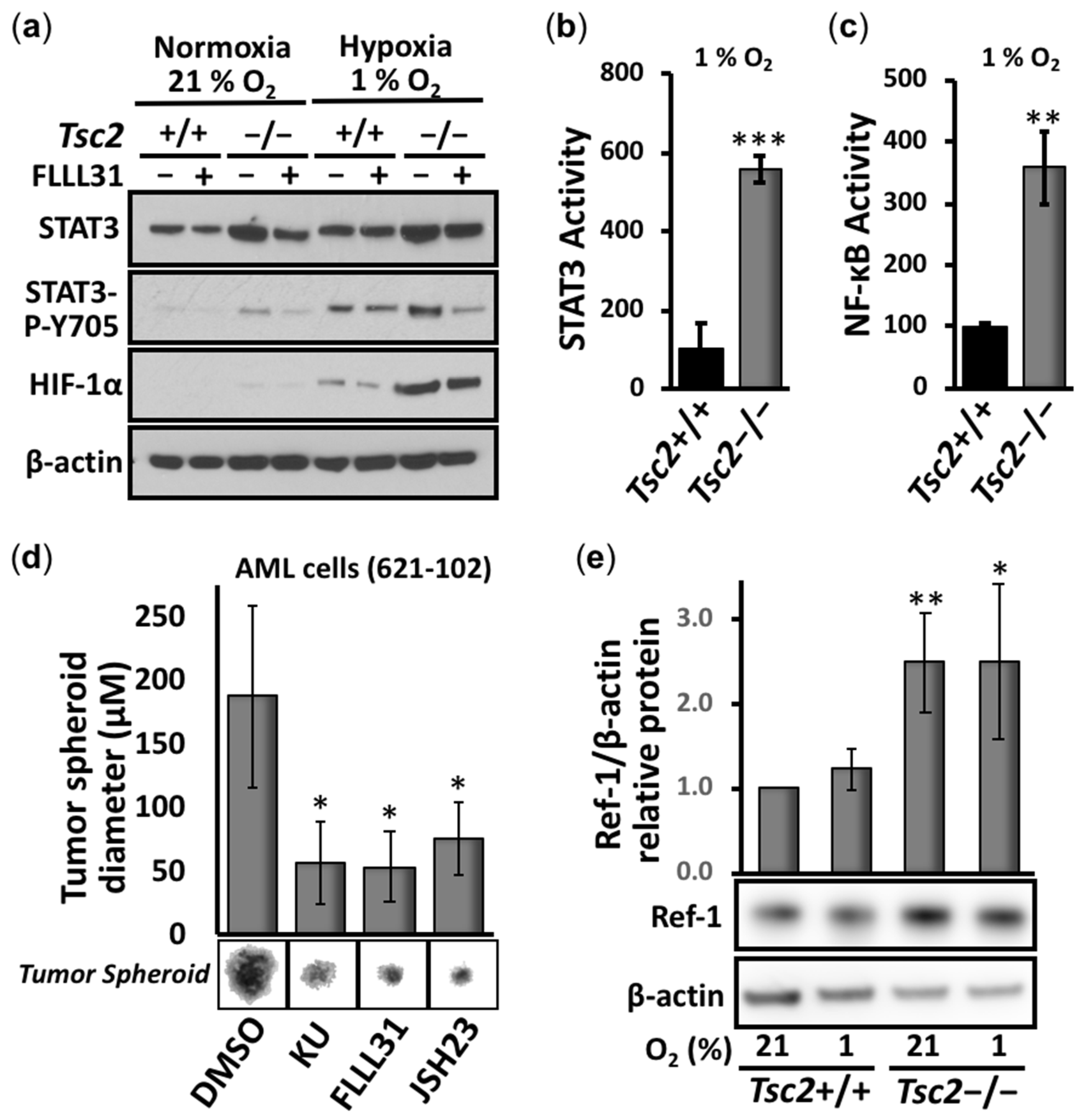
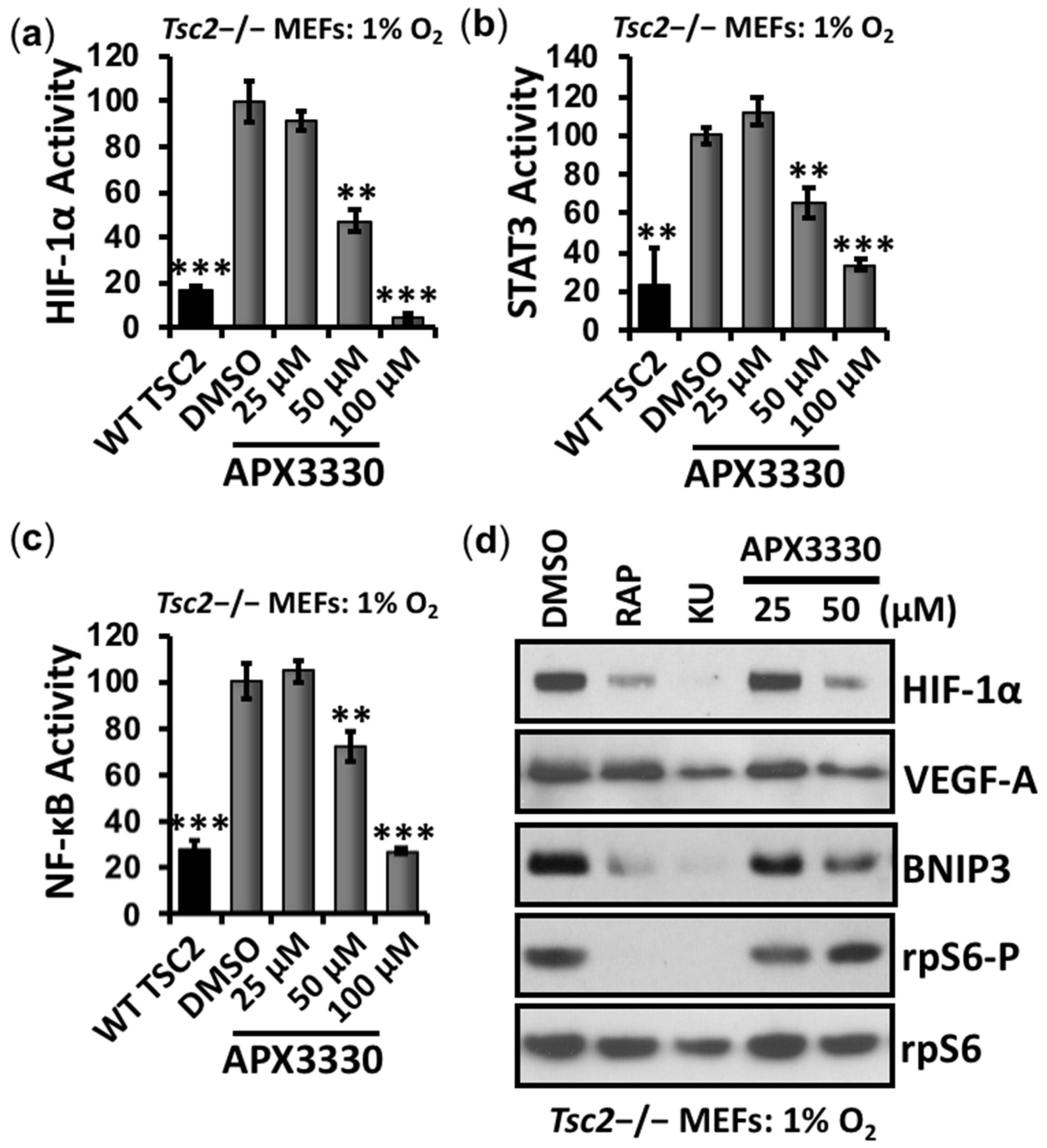
3.1. Aberrant Activation of HIF-1α/STAT3/NF-κB in Tsc2-Deficient Cells Is Restored with APX3330, a Ref-1-Specific Inhibitor
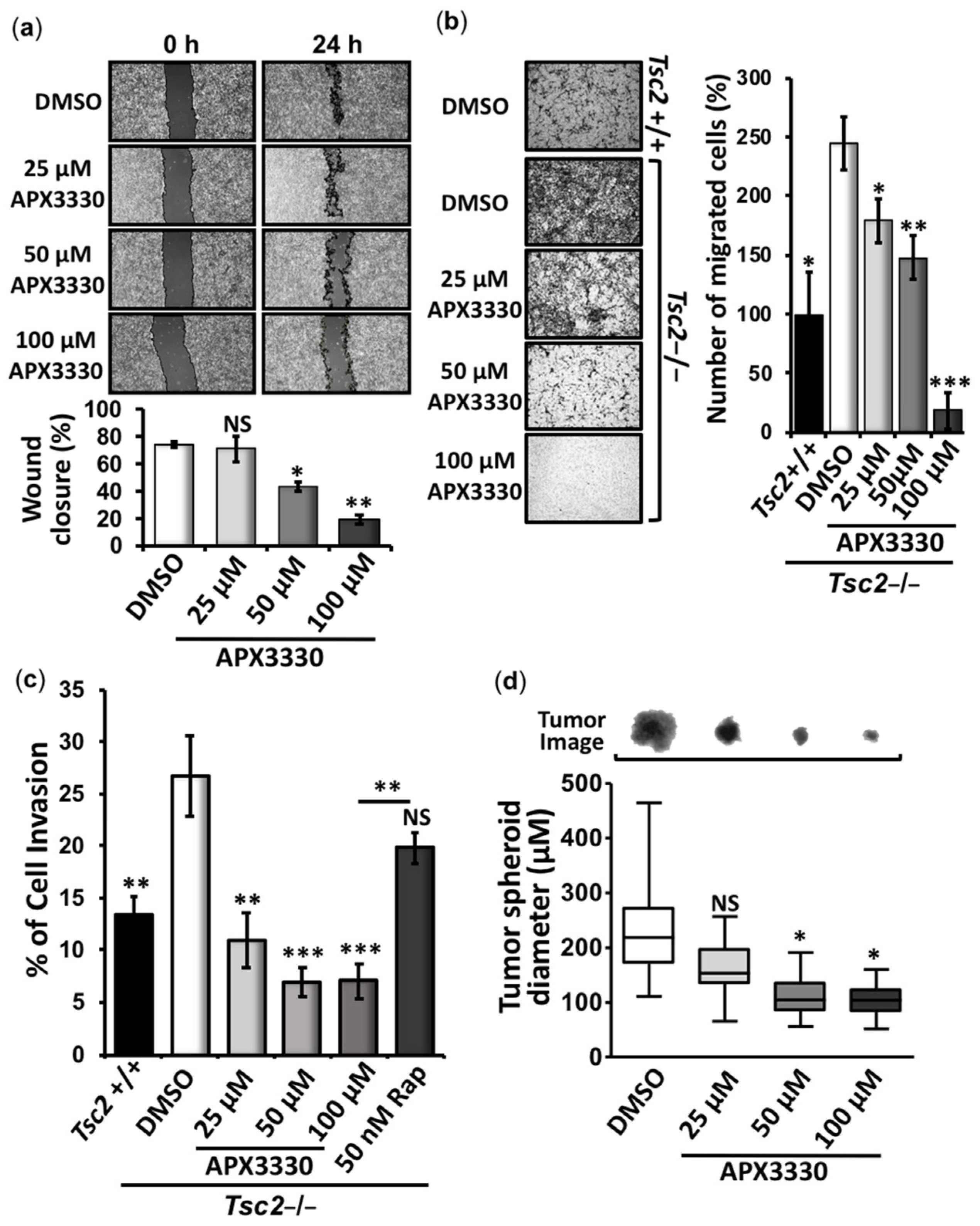
3.2. APX3330 Inhibits Cell Migration/Invasion and Tumor Spheroid Formation of Tsc2−/− MEFs
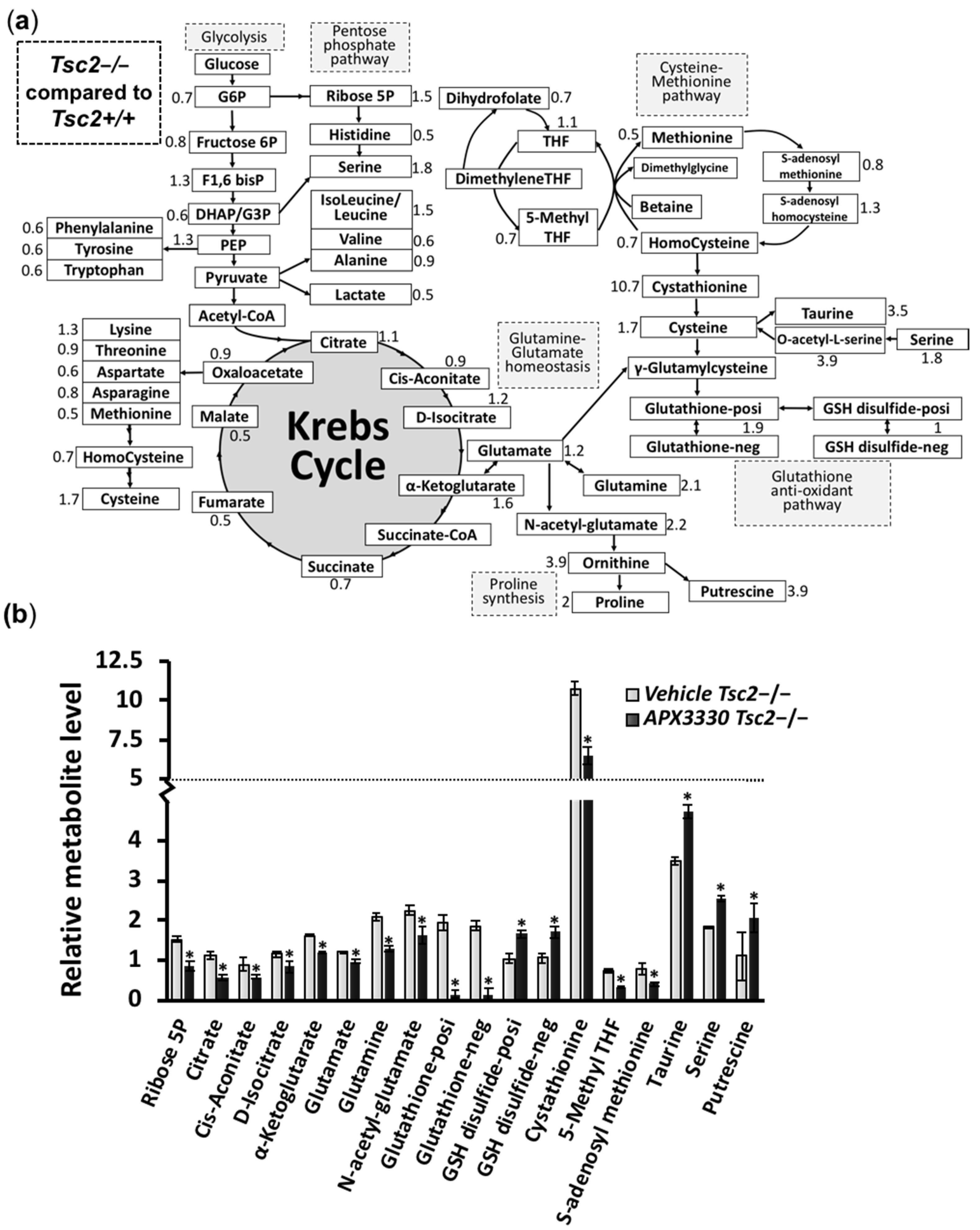
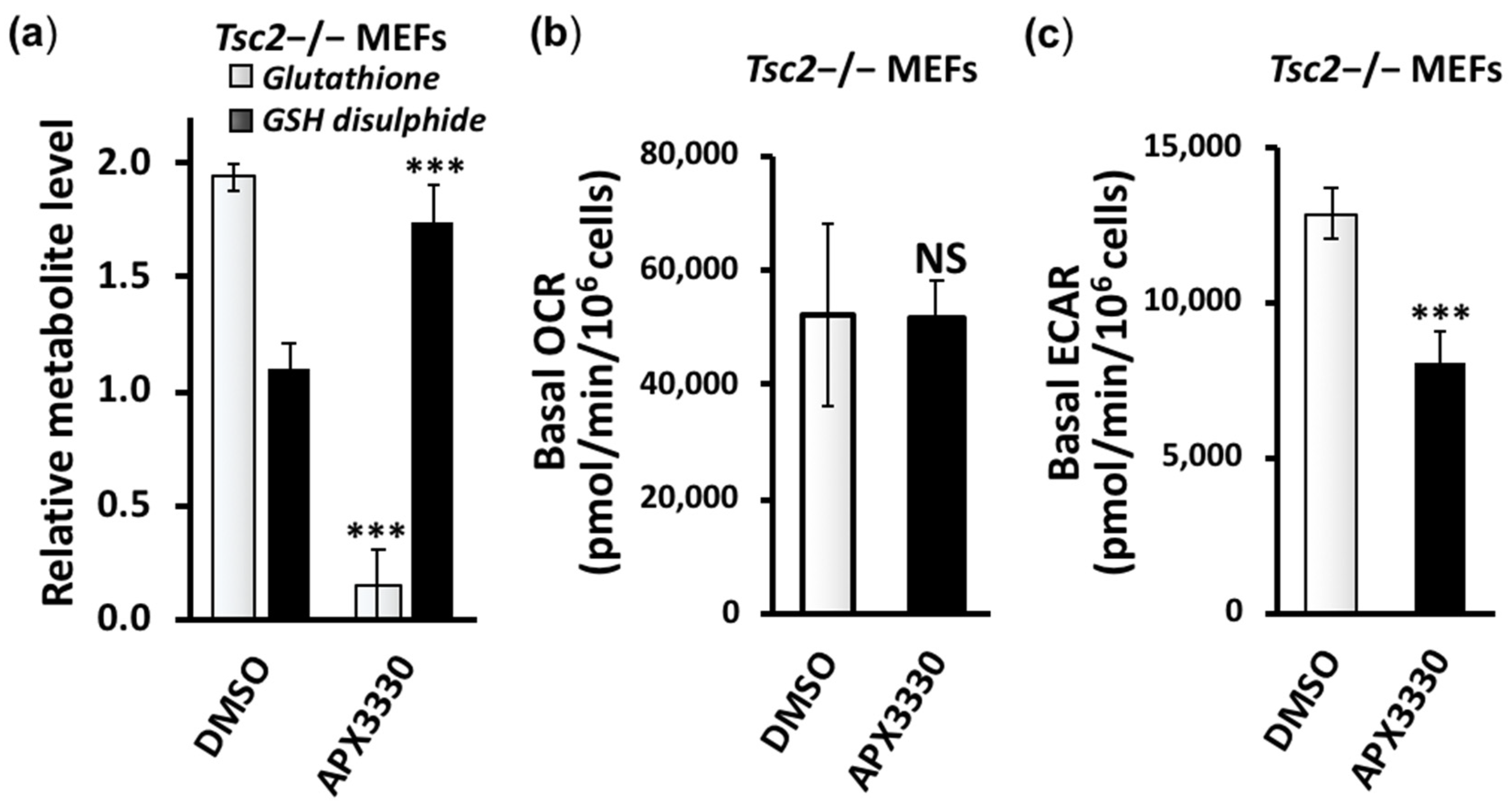
3.3. Metabolic Profiling Shows Restoration of Redox Homeostasis in the Tsc2−/− MEFs after APX3330 Treatment
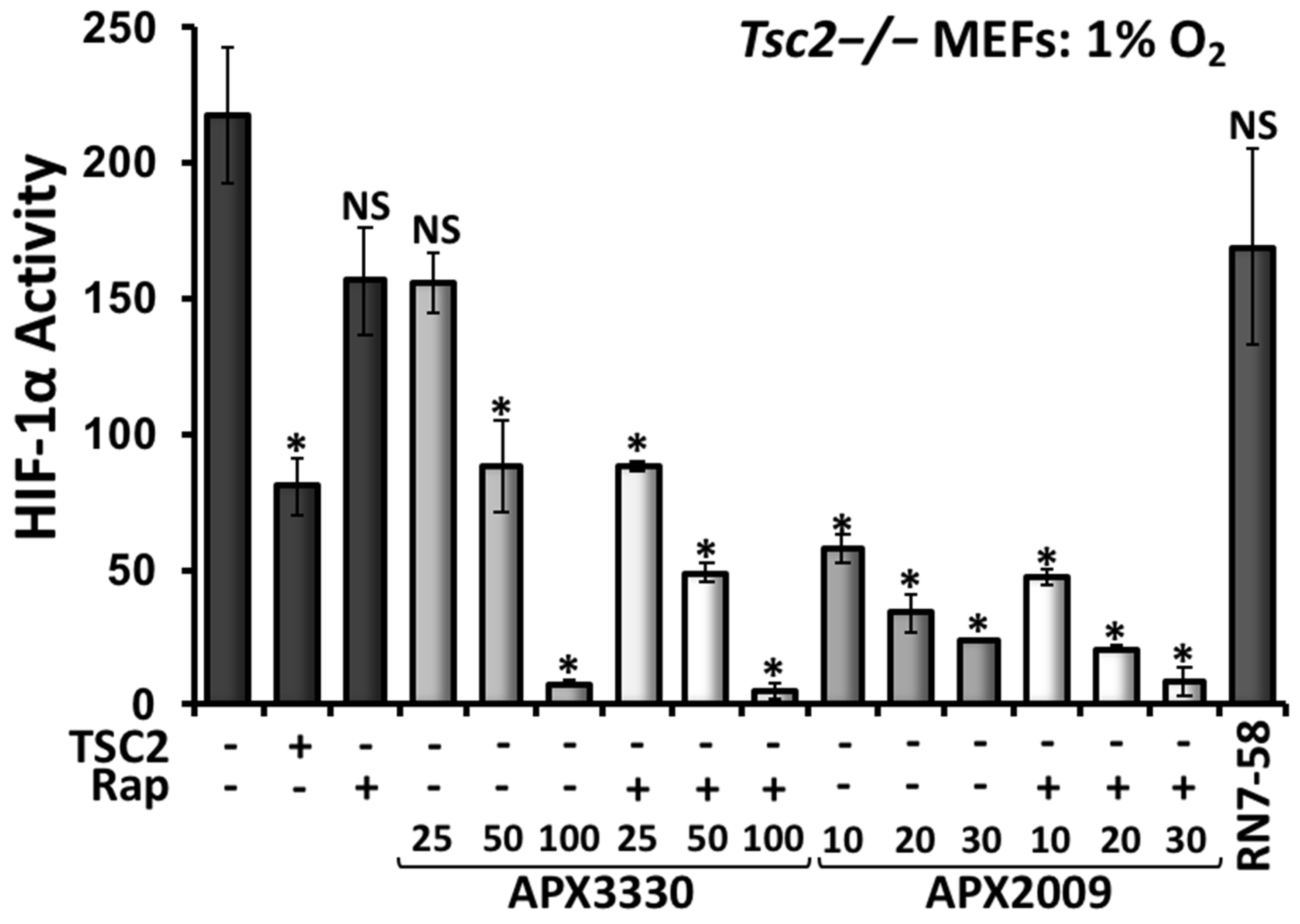
3.4. Second-Generation Ref-1 Inhibitor APX2009 Shows Increased Potency to Inhibit HIF-1α
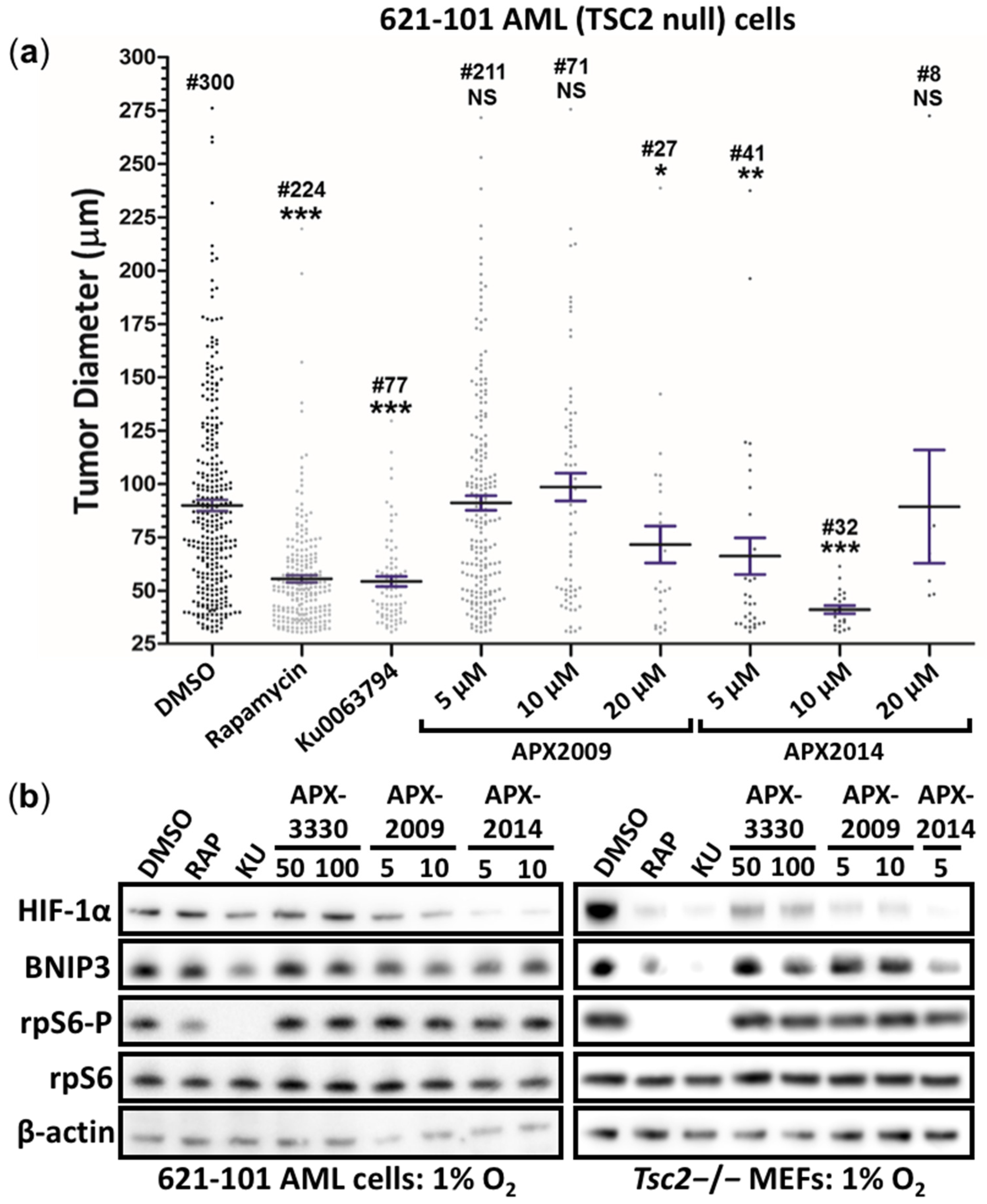
3.5. Second-Generation Ref-1 Inhibitors APX2009 and APX2014 Inhibit In Vitro Tumor Growth of AML (621-102) Cells
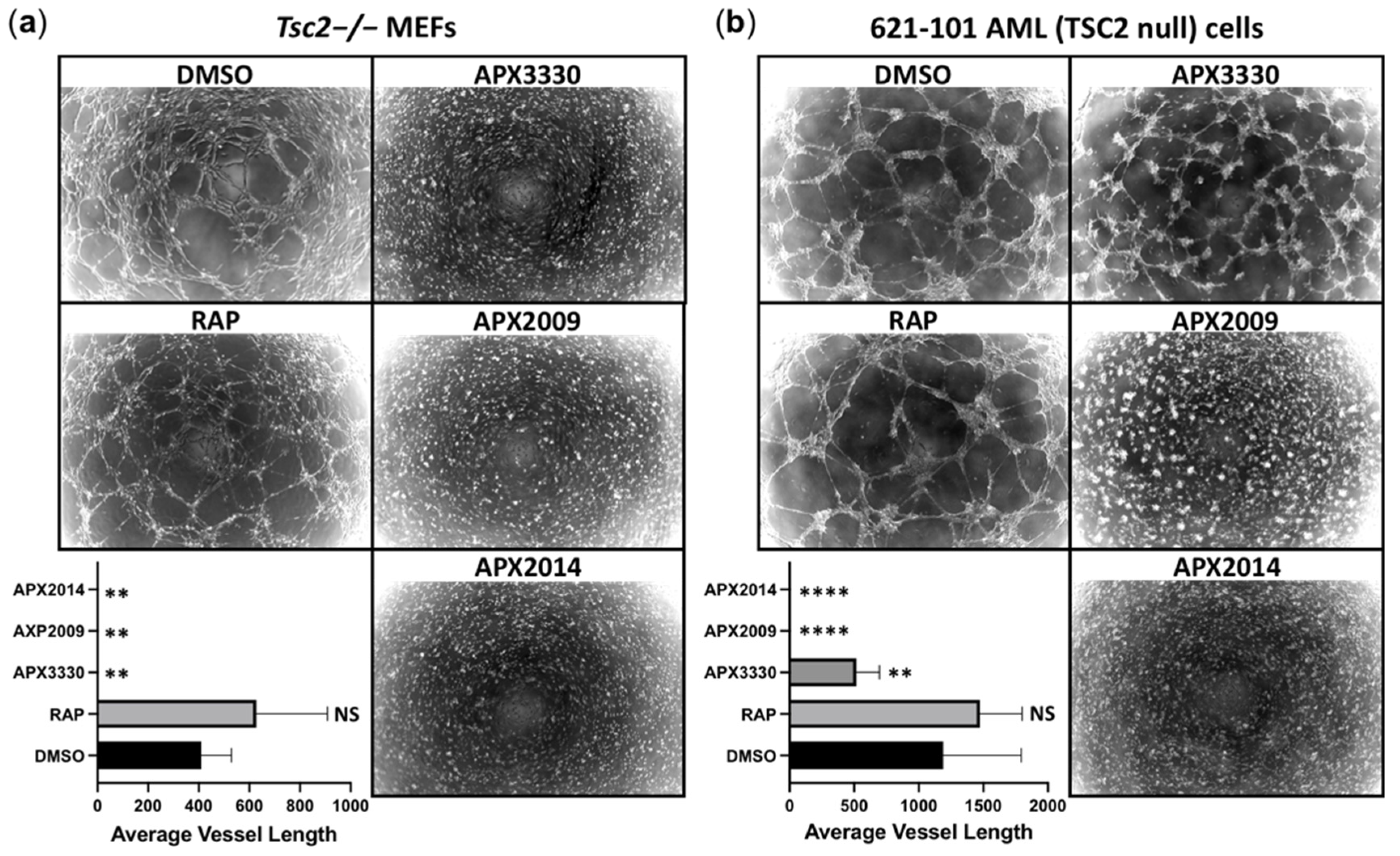
3.6. Vasculature Mimicry Was Blocked with APX3330, APX2009, and APX2014, While mTOR Inhibition Was Ineffective
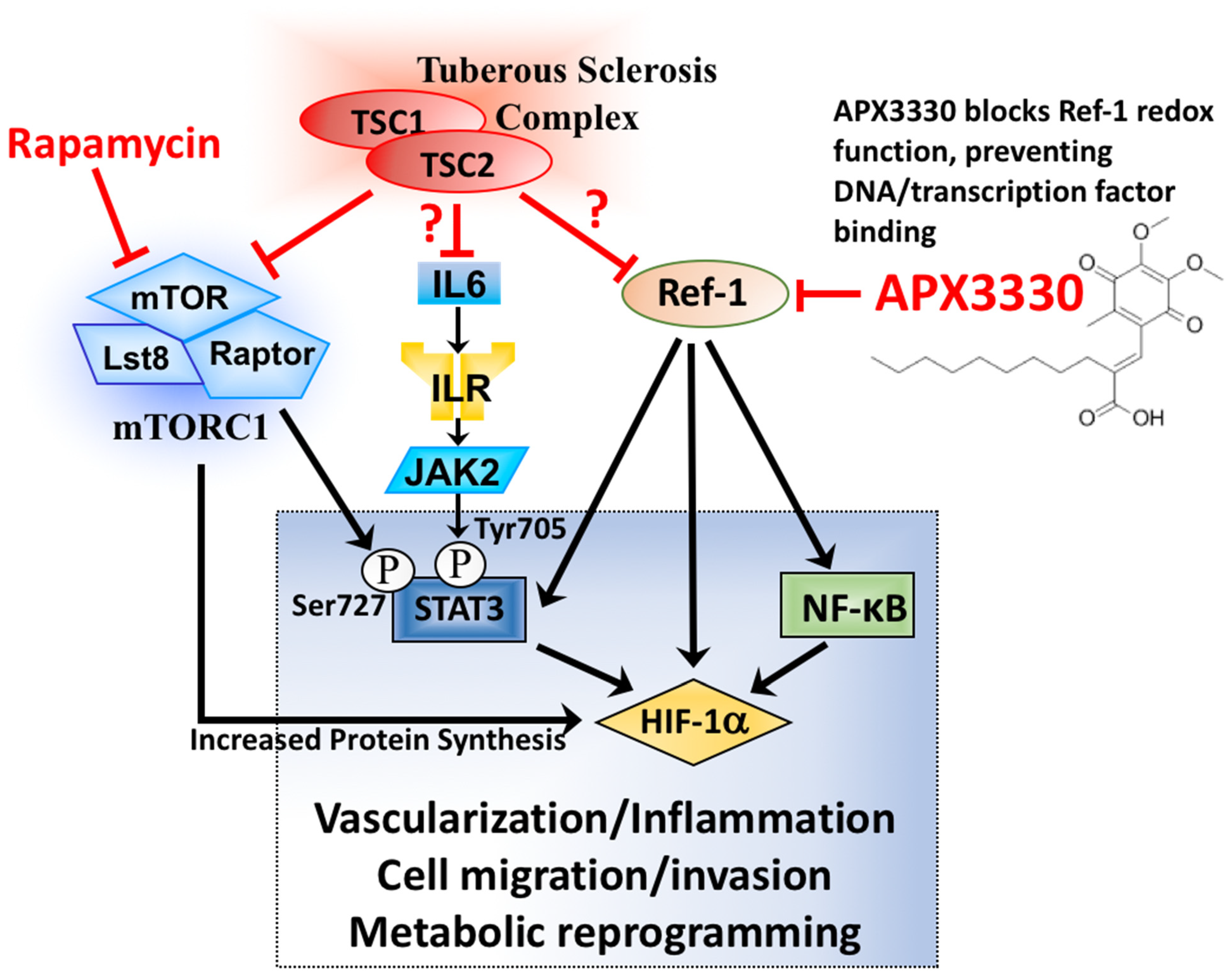
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McEneaney, L.J.; Tee, A.R. Finding a cure for tuberous sclerosis complex: From genetics through to targeted drug therapies. In Advances in Genetics; Kumar, D., Ed.; Elsevier: New York, NY, USA, 2019; Volume 103, pp. 91–118. [Google Scholar] [CrossRef]
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Pirson, Y. Tuberous sclerosis complex-associated kidney angiomyolipoma: From contemplation to action. Nephrol. Dial. Transpl. 2013, 28, 1680–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Fallah, A. Optimal management of seizures associated with tuberous sclerosis complex: Current and emerging options. Neuropsychiatr. Dis. Treat. 2014, 10, 2021–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henske, E.P.; McCormack, F.X. Lymphangioleiomyomatosis—A wolf in sheep’s clothing. J. Clin. Investig. 2012, 122, 3807–3816. [Google Scholar] [CrossRef] [Green Version]
- Carsillo, T.; Astrinidis, A.; Henske, E.P. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2000, 97, 6085–6090. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Rad, E.; Murray, J.T.; Tee, A.R. Oncogenic Signalling through Mechanistic Target of Rapamycin (mTOR): A Driver of Metabolic Transformation and Cancer Progression. Cancers 2018, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [Green Version]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; Garcìa-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himes, B.E.; Obraztsova, K.; Lian, L.; Shumyatcher, M.; Rue, R.; Atochina-Vasserman, E.N.; Hur, S.K.; Bartolomei, M.S.; Evans, J.F.; Krymskaya, V.P. Rapamycin-independent IGF2 expression in Tsc2-null mouse embryo fibroblasts and human lymphangioleiomyomatosis cells. PLoS ONE 2018, 13, e0197105. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M.; et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijit, M.; Caston, R.; Gampala, S.; Fishel, M.L.; Fehrenbacher, J.; Kelley, M.R. APE1/Ref-1—One Target with Multiple Indications: Emerging Aspects and New Directions. J. Cell. Signal. 2021, 2, 151–161. [Google Scholar]
- Yu, J.; Astrinidis, A.; Howard, S.; Henske, E.P. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L694–L700. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Johnson, C.E.; Wiltshire, M.; Errington, R.J.; Tee, A.R. Targeting protein homeostasis with nelfinavir/salinomycin dual therapy effectively induces death of mTORC1 hyperactive cells. Oncotarget 2017, 8, 48711–48724. [Google Scholar] [CrossRef] [Green Version]
- Parkhitko, A.A.; Priolo, C.; Coloff, J.L.; Yun, J.; Wu, J.J.; Mizumura, K.; Xu, W.; Malinowska, I.A.; Yu, J.; Kwiatkowski, D.J.; et al. Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Mol. Cancer Res. 2014, 12, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef]
- Tanaka, H.; Arima, M. tuberous sclerosis: Hydroxyproline content in urine and tissues. Brain Dev. 1981, 3, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nakazawa, K.; Arima, M.; Morooka, K.; Suzuki, F.; Aoki, T.; Kohno, Y. Tuberous sclerosis: Proline and hydroxyproline contents in serum. Brain Dev. 1983, 5, 450–456. [Google Scholar] [CrossRef]
- Gampala, S.; Shah, F.; Lu, X.; Moon, H.R.; Babb, O.; Umesh Ganesh, N.; Sandusky, G.; Hulsey, E.; Armstrong, L.; Mosely, A.L.; et al. Ref-1 redox activity alters cancer cell metabolism in pancreatic cancer: Exploiting this novel finding as a potential target. J. Exp. Clin. Cancer Res. 2021, 40, 251. [Google Scholar] [CrossRef]
- Ando, K.; Hirao, S.; Kabe, Y.; Ogura, Y.; Sato, I.; Yamaguchi, Y.; Wada, T.; Handa, H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008, 36, 4327–4336. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1α under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.W.; Fishel, M.L.; Boos, A.; Kelley, M.R.; Jerde, T.J. APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells. Oncotarget 2017, 9, 10962–10977. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Luo, M.; Reed, A.; Su, D.; Delaplane, S.; Borch, R.F.; Nyland, R.L.; Gross, M.L.; Georgiadis, M.M. Functional analysis of novel analogues of E3330 that block the redox signaling activity of the multifunctional AP endonuclease/redox signaling enzyme APE1/Ref-1. Antioxid. Redox Signal. 2011, 14, 1387–1401. [Google Scholar] [CrossRef]
- Jiang, A.; Gao, H.; Kelley, M.R.; Qiao, X. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vis. Res. 2011, 51, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.A.; Gao, H.; Qiao, X. Suppression of choroidal neovascularization through inhibition of APE1/Ref-1 redox activity. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4461–4469. [Google Scholar] [CrossRef]
- Sardar Pasha, S.; Sishtla, K.; Sulaiman, R.S.; Park, B.; Shetty, T.; Shah, F.; Fishel, M.L.; Wikel, J.H.; Kelley, M.R.; Corson, T.W. Ref-1/APE1 Inhibition with Novel Small Molecules Blocks Ocular Neovascularization. J. Pharmacol. Exp. Ther. 2018, 367, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. BioMed Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Anderson, A.K.L.; Landers, M.A.; Wang, Y.; Kelley, M.R.; Messmann, R.A. CTC enumeration and characterization as a pharmacodynamic marker in the phase I clinical study of APX3330, an APE1/Ref-1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, e14531. [Google Scholar] [CrossRef]
- Shahda, S.; Lakhani, N.J.; O’Neil, B.; Rasco, D.W.; Wan, J.; Mosley, A.L.; Liu, H.; Kelley, M.R.; Messmann, R.A. A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3097. [Google Scholar] [CrossRef]
- Fishel, M.L.; Xia, H.; McGeown, J.; McIlwain, D.W.; Elbanna, M.; Craft, A.A.; Kaimakliotis, H.Z.; Sandusky, G.E.; Zhang, C.; Pili, R.; et al. Antitumor Activity and Mechanistic Characterization of APE1/Ref-1 Inhibitors in Bladder Cancer. Mol. Cancer Ther. 2019, 18, 1947–1960. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Wikel, J.H.; Guo, C.; Pollok, K.E.; Bailey, B.J.; Wireman, R.; Fishel, M.L.; Vasko, M.R. Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy-Induced Peripheral Neuropathy. J. Pharmacol. Exp. Ther. 2016, 359, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.H.; Bero, A.W.; Zhang, B.; Holtzman, D.M.; Wong, M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of Tuberous Sclerosis Complex. Neurobiol. Dis. 2010, 37, 764–771. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Champion, J.D.; Dodd, K.M.; Lam, H.C.; Alzahrani, M.A.M.; Seifan, S.; Rad, E.; Scourfield, D.O.; Fishel, M.L.; Calver, B.L.; Ager, A.; et al. Drug Inhibition of Redox Factor-1 Restores Hypoxia-Driven Changes in Tuberous Sclerosis Complex 2 Deficient Cells. Cancers 2022, 14, 6195. https://doi.org/10.3390/cancers14246195
Champion JD, Dodd KM, Lam HC, Alzahrani MAM, Seifan S, Rad E, Scourfield DO, Fishel ML, Calver BL, Ager A, et al. Drug Inhibition of Redox Factor-1 Restores Hypoxia-Driven Changes in Tuberous Sclerosis Complex 2 Deficient Cells. Cancers. 2022; 14(24):6195. https://doi.org/10.3390/cancers14246195
Chicago/Turabian StyleChampion, Jesse D., Kayleigh M. Dodd, Hilaire C. Lam, Mohammad A. M. Alzahrani, Sara Seifan, Ellie Rad, David Oliver Scourfield, Melissa L. Fishel, Brian L. Calver, Ann Ager, and et al. 2022. "Drug Inhibition of Redox Factor-1 Restores Hypoxia-Driven Changes in Tuberous Sclerosis Complex 2 Deficient Cells" Cancers 14, no. 24: 6195. https://doi.org/10.3390/cancers14246195