
GMEB2 Promotes the Growth of Colorectal Cancer by Activating ADRM1 Transcription and NF-κB Signalling and Is Positively Regulated by the m6A Reader YTHDF1
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Databases
2.2. Cell Lines and Cell Culture
2.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR) Analysis
2.4. Western Blotting
2.5. Immunohistochemistry (IHC) Staining
2.6. Plasmid Construction, Cell Transfection and Lentivirus Vector Infection
2.7. Cell Proliferation and Colony Formation Assay
2.8. Tumor Xenografts
2.9. Chromatin Immunoprecipitation (ChIP) Assay
2.10. Dual-Luciferase Reporter Assay
2.11. Immunofluorescent Staining
2.12. Methylated RNA Immunoprecipitation (MeRIP)
2.13. RNA Immunoprecipitation (RIP) Assay
2.14. Statistical Analysis
3. Results
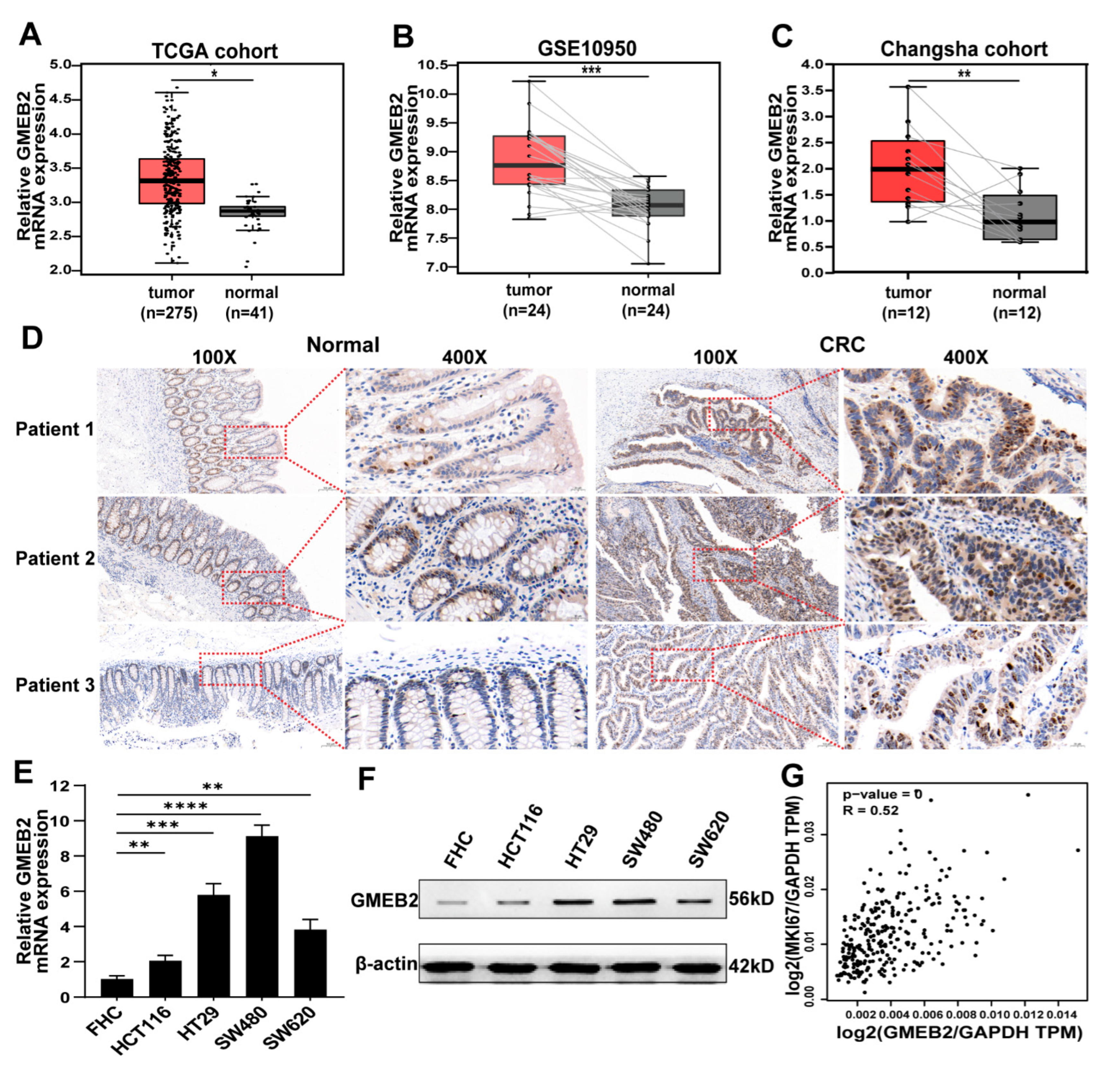
3.1. GMEB2 Expression Is Elevated in CRC
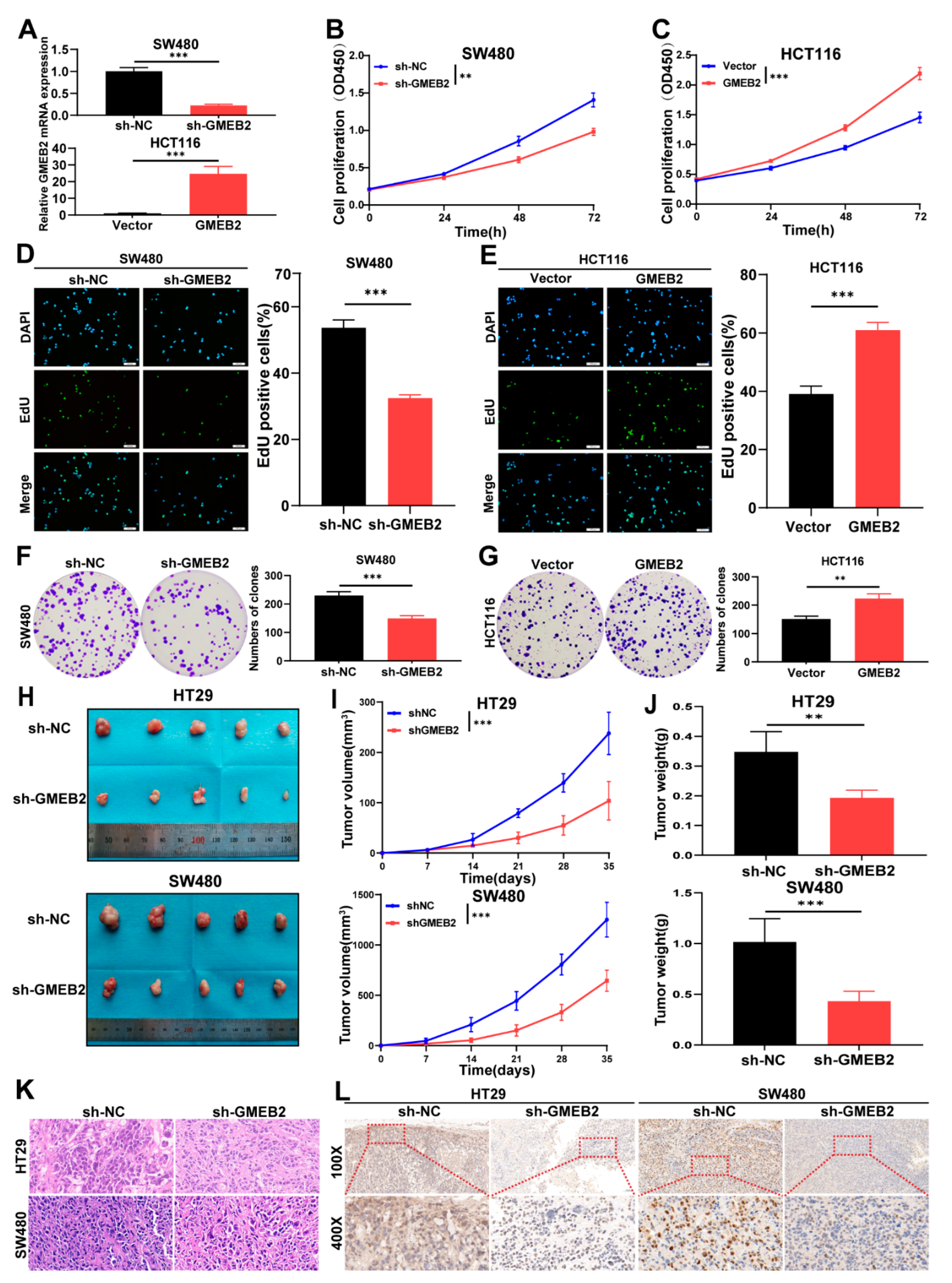
3.2. GMEB2 Drives CRC Cell Growth In Vitro and In Vivo
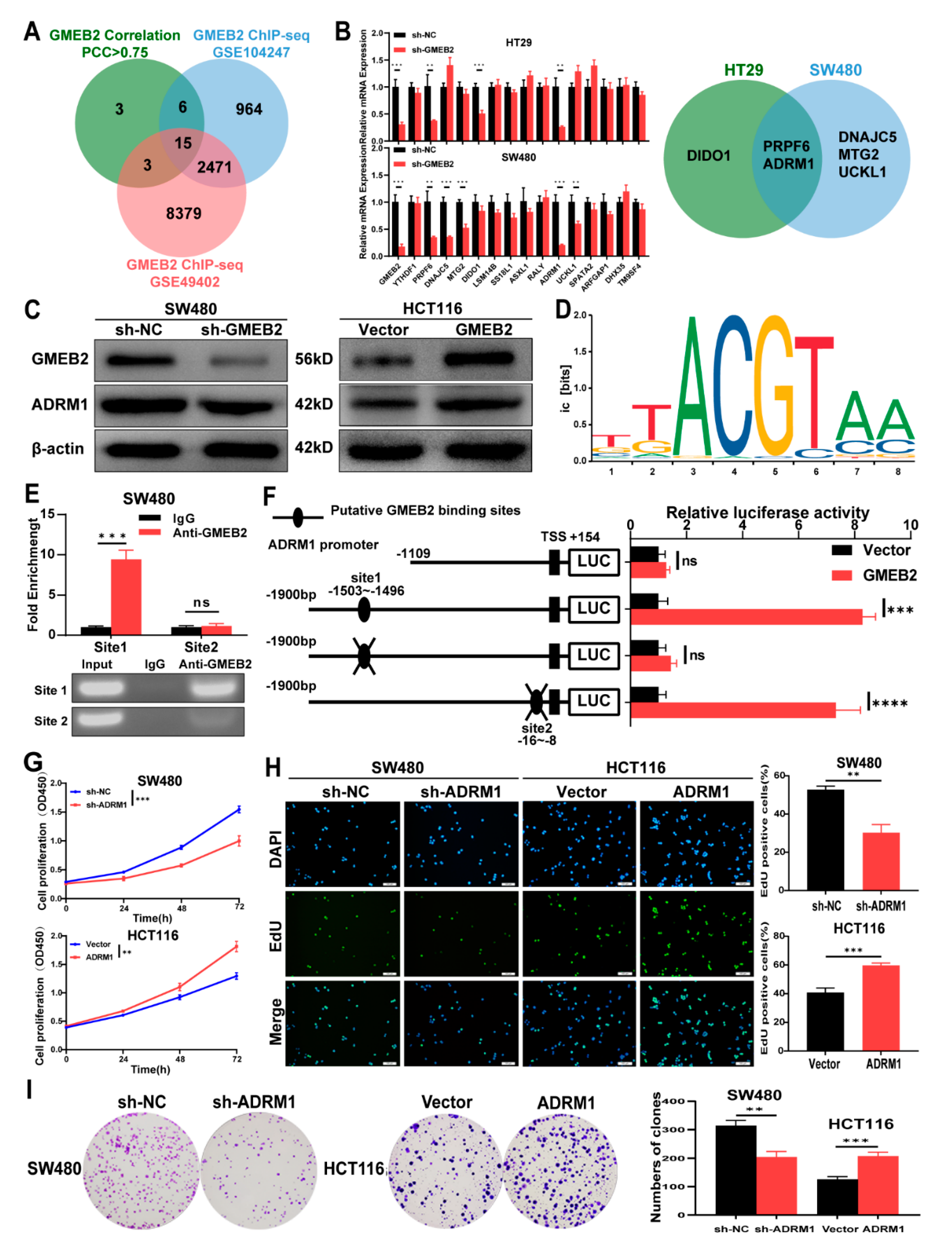
3.3. ADRM1 Is Transactivated by GMEB2, and Promotes CRC Cell Growth
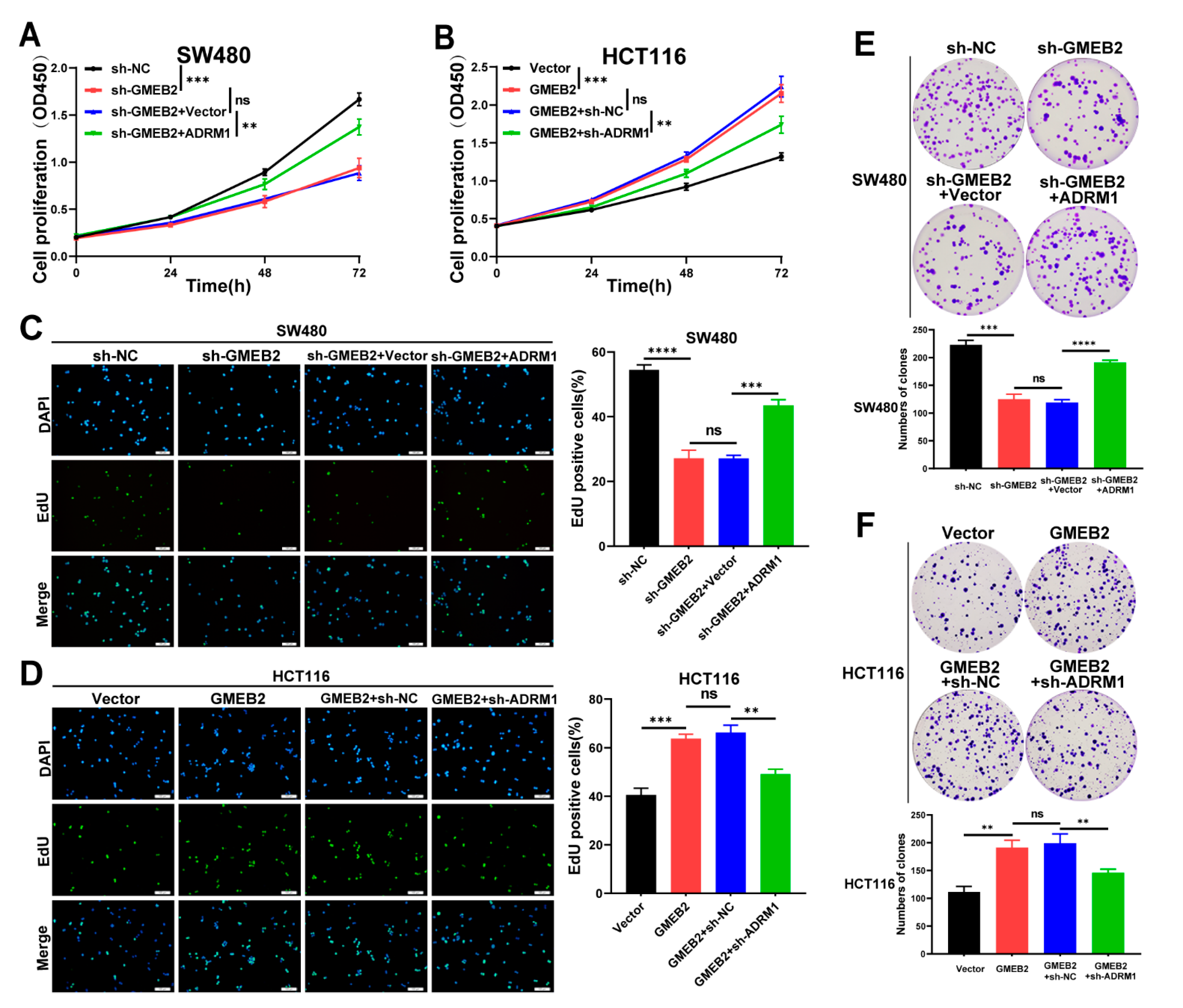
3.4. The GMEB2/ADRM1 Axis Promotes CRC Cell Growth In Vitro
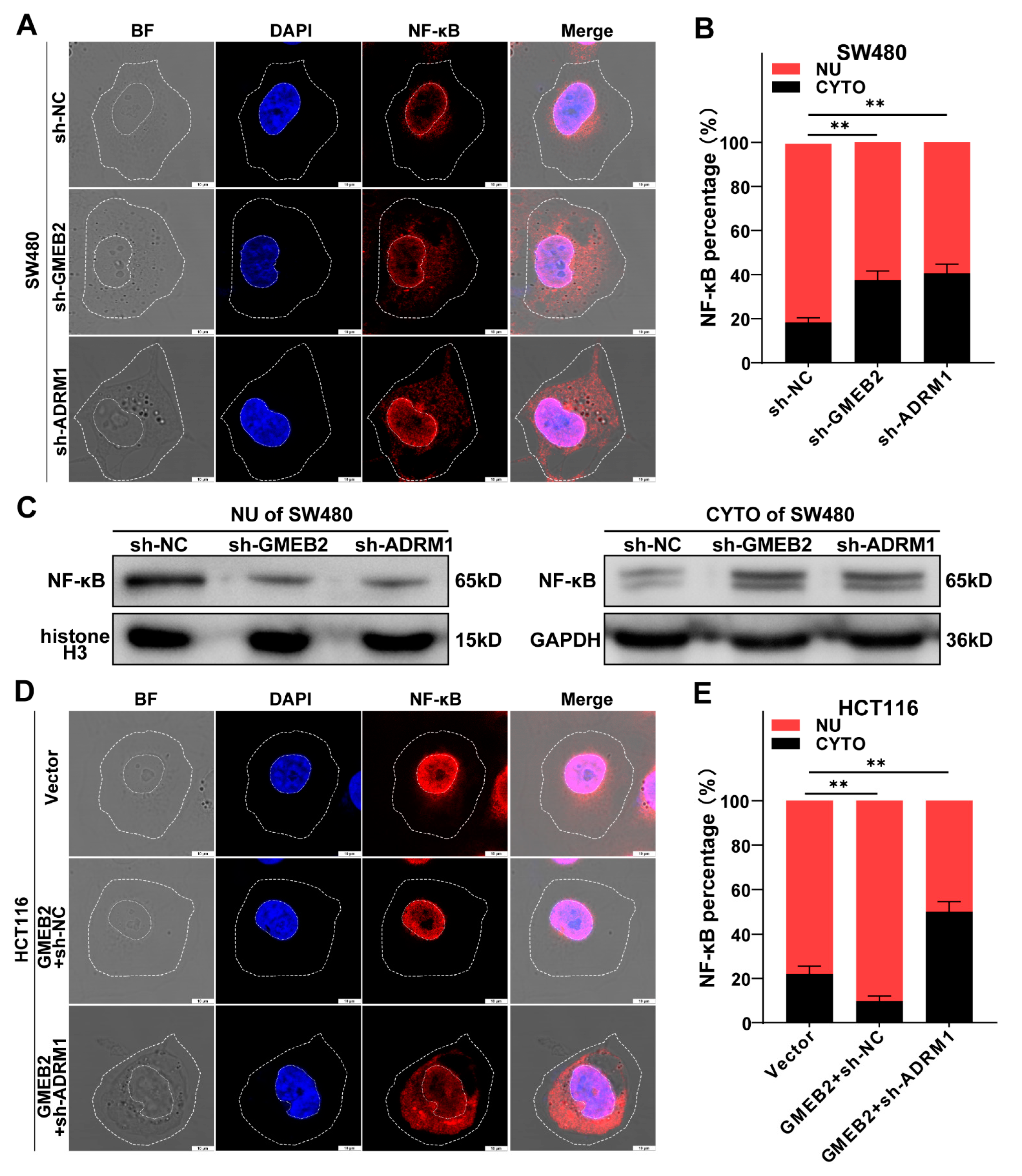
3.5. The GMEB2/ADRM1 Axis Promotes Nuclear Translocation of NF-κB to Activate NF-κB Signalling
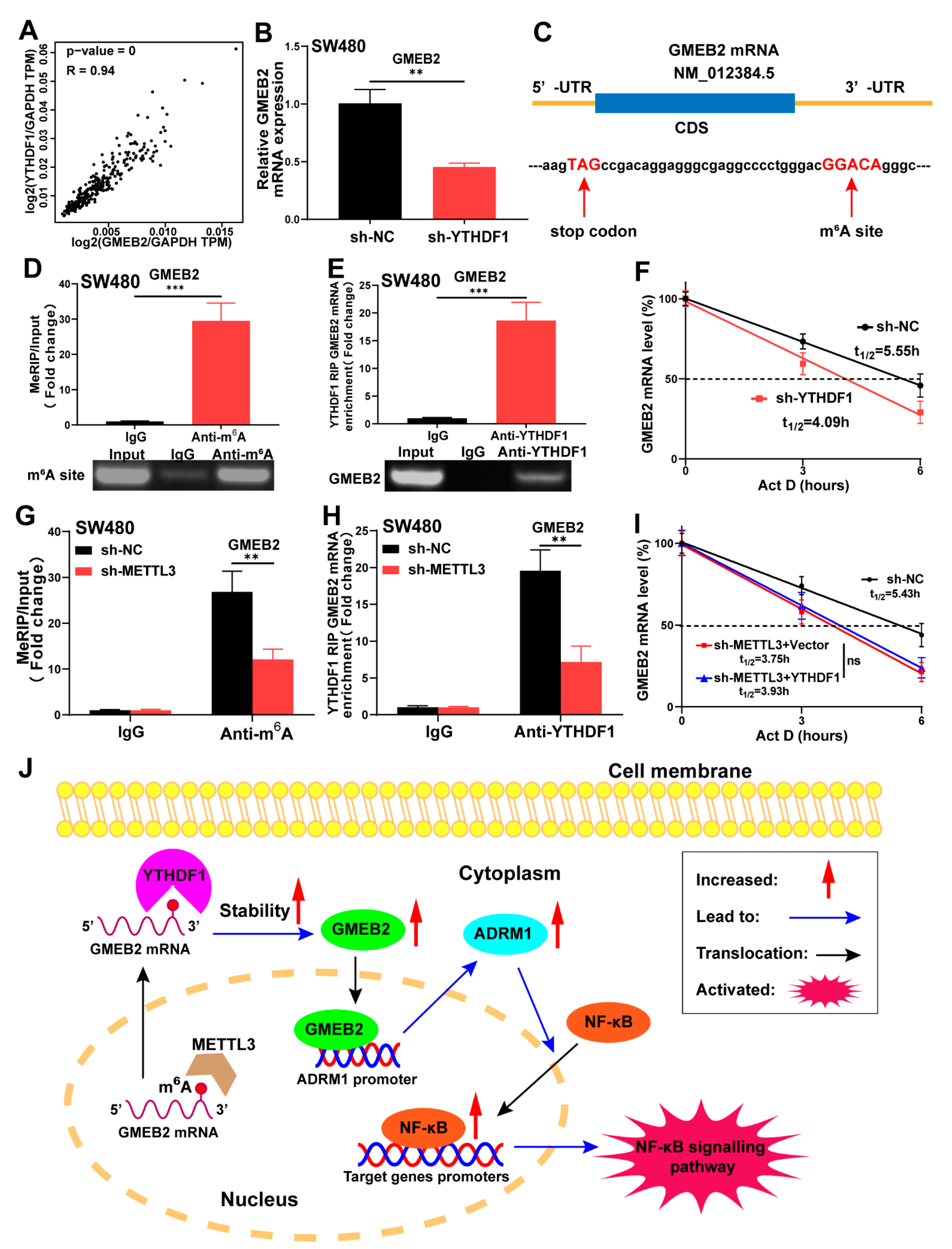
3.6. GMEB2 mRNA Is Stabilized by YTHDF1 in an m6A-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar]
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 135, 584–590. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar]
- Zeng, H.; Plisov, S.Y.; Simons, S.S., Jr. Ability of the glucocorticoid modulatory element to modify glucocorticoid receptor transactivation indicates parallel pathways for the expression of glucocorticoid modulatory element and glucocorticoid response element activities. Mol. Cell. Endocrinol. 2000, 162, 221–234. [Google Scholar]
- Chen, J.; He, Y.; Simons, S.S., Jr. Structure/activity relationships for GMEB-2: The second member of the glucocorticoid modulatory element-binding complex. Biochemistry 2004, 43, 245–255. [Google Scholar]
- Zeng, H.; Kaul, S.; Simons, S.S., Jr. Genomic organization of human GMEB-1 and rat GMEB-2: Structural conservation of two multifunctional proteins. Nucleic Acids Res. 2000, 28, 1819–1829. [Google Scholar]
- Kaul, S.; Blackford, J.A., Jr.; Chen, J.; Ogryzko, V.V.; Simons, S.S., Jr. Properties of the glucocorticoid modulatory element binding proteins GMEB-1 and -2: Potential new modifiers of glucocorticoid receptor transactivation and members of the family of KDWK proteins. Mol. Endocrinol. 2000, 14, 1010–1027. [Google Scholar]
- Cui, Z.; Sun, Q.; Yan, W.; Han, Q.; Wang, G.; Hu, Y. The role of miR-320a and its target gene GMEB1 in epithelial-mesenchymal transition and invasion of colorectal cancer. J. Gene Med. 2021, e3327. [Google Scholar] [CrossRef]
- Kotsaris, G.; Kerselidou, D.; Koutsoubaris, D.; Constantinou, E.; Malamas, G.; Garyfallos, D.A.; Hatzivassiliou, E.G. TRAF3 can interact with GMEB1 and modulate its anti-apoptotic function. J. Biol. Res. 2020, 27, 7. [Google Scholar]
- An, W.; Yao, S.; Sun, X.; Hou, Z.; Lin, Y.; Su, L.; Liu, X. Glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the de-ubiquitinase USP40 to stabilize CFLAR(L) and inhibit apoptosis in human non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 181. [Google Scholar]
- Nakagawa, T.; Tsuruma, K.; Uehara, T.; Nomura, Y. GMEB1, a novel endogenous caspase inhibitor, prevents hypoxia- and oxidative stress-induced neuronal apoptosis. Neurosci. Lett. 2008, 438, 34–37. [Google Scholar]
- Tsuruma, K.; Nakagawa, T.; Morimoto, N.; Minami, M.; Hara, H.; Uehara, T.; Nomura, Y. Glucocorticoid modulatory element-binding protein 1 binds to initiator procaspases and inhibits ischemia-induced apoptosis and neuronal injury. J. Biol. Chem. 2006, 281, 11397–11404. [Google Scholar]
- Tsuruma, K.; Nakagawa, T.; Shirakura, H.; Hayashi, N.; Uehara, T.; Nomura, Y. Regulation of procaspase-2 by glucocorticoid modulatory element-binding protein 1 through the interaction with caspase recruitment domain. Biochem. Biophys. Res. Commun. 2004, 325, 1246–1251. [Google Scholar]
- Pi, J.; Wang, W.; Ji, M.; Wang, X.; Wei, X.; Jin, J.; Liu, T.; Qiang, J.; Qi, Z.; Li, F.; et al. YTHDF1 Promotes Gastric Carcinogenesis by Controlling Translation of FZD7. Cancer Res. 2021, 81, 2651–2665. [Google Scholar]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target Ther. 2021, 6, 74. [Google Scholar]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar]
- Chen, Z.; Zhong, X.; Xia, M.; Zhong, J. The roles and mechanisms of the m6A reader protein YTHDF1 in tumor biology and human diseases. Mol. Ther. Nucleic Acids 2021, 26, 1270–1279. [Google Scholar]
- He, M.; Jiang, D.; Xun, A.; Yang, J.; Luo, Q.; Wu, H. METTL3 enhances PNN mRNA stability through m6A modification to augment tumorigenesis of colon adenocarcinoma. Exp. Physiol. 2022. [Google Scholar]
- Song, Y.; Du, T.; Ray, A.; Chauhan, K.; Samur, M.; Munshi, N.; Chauhan, D.; Anderson, K.C. Identification of novel anti-tumor therapeutic target via proteomic characterization of ubiquitin receptor ADRM1/Rpn13. Blood Cancer J. 2021, 11, 13. [Google Scholar]
- Pilarsky, C.; Wenzig, M.; Specht, T.; Saeger, H.D.; Grützmann, R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 2004, 6, 744–750. [Google Scholar]
- Chen, W.; Hu, X.T.; Shi, Q.L.; Zhang, F.B.; He, C. Silencing of Adrm1 by RNA interference suppresses proliferation of colorectal cancer cells. Zhonghua Zhong Liu Za Zhi 2009, 31, 815–819. [Google Scholar]
- Chen, W.; Hu, X.T.; Shi, Q.L.; Zhang, F.B.; He, C. Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer cell migration, survival and tumorigenicity. Oncol. Rep. 2009, 21, 531–537. [Google Scholar]
- Carvalho, B.; Postma, C.; Mongera, S.; Hopmans, E.; Diskin, S.; van de Wiel, M.A.; van Criekinge, W.; Thas, O.; Matthäi, A.; Cuesta, M.A.; et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut 2009, 58, 79–89. [Google Scholar]
- Liang, Y.C.; Wang, J.L.; Wang, H.T.; Liu, H.; Zhang, H.L.; Liang, Y.X. ADRM1 as a therapeutic target in hepatocellular carcinoma. Kaohsiung J. Med. Sci. 2021, 37, 47–54. [Google Scholar]
- Wang, J.; Wu, X.; Dai, W.; Li, J.; Xiang, L.; Tang, W.; Lin, J.; Zhang, W.; Liu, G.; Yang, Q.; et al. The CCDC43-ADRM1 axis regulated by YY1, promotes proliferation and metastasis of gastric cancer. Cancer Lett. 2020, 482, 90–101. [Google Scholar]
- Fejzo, M.S.; Anderson, L.; Chen, H.W.; Anghel, A.; Zhuo, J.; Anchoori, R.; Roden, R.; Slamon, D.J. ADRM1-amplified metastasis gene in gastric cancer. Genes Chromosomes Cancer 2015, 54, 506–515. [Google Scholar]
- Jang, S.H.; Park, J.W.; Kim, H.R.; Seong, J.K.; Kim, H.K. ADRM1 gene amplification is a candidate driver for metastatic gastric cancers. Clin. Exp. Metastasis 2014, 31, 727–733. [Google Scholar]
- Jiang, R.T.; Yemelyanova, A.; Xing, D.; Anchoori, R.K.; Hamazaki, J.; Murata, S.; Seidman, J.D.; Wang, T.L.; Roden, R.B.S. Early and consistent overexpression of ADRM1 in ovarian high-grade serous carcinoma. J. Ovarian Res. 2017, 10, 53. [Google Scholar]
- Fejzo, M.S.; Anderson, L.; von Euw, E.M.; Kalous, O.; Avliyakulov, N.K.; Haykinson, M.J.; Konecny, G.E.; Finn, R.S.; Slamon, D.J. Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 3094–3109. [Google Scholar]
- Soong, R.S.; Anchoori, R.K.; Roden, R.B.S.; Cho, R.L.; Chen, Y.C.; Tseng, S.C.; Huang, Y.L.; Liao, P.C.; Shyu, Y.C. Bis-benzylidine Piperidone RA190 treatment of hepatocellular carcinoma via binding RPN13 and inhibiting NF-κB signaling. BMC Cancer 2020, 20, 386. [Google Scholar]
- Yu, G.Y.; Wang, X.; Zheng, S.S.; Gao, X.M.; Jia, Q.A.; Zhu, W.W.; Lu, L.; Jia, H.L.; Chen, J.H.; Dong, Q.Z.; et al. RA190, a Proteasome Subunit ADRM1 Inhibitor, Suppresses Intrahepatic Cholangiocarcinoma by Inducing NF-KB-Mediated Cell Apoptosis. Cell Physiol. Biochem. 2018, 47, 1152–1166. [Google Scholar]
- Anchoori, R.K.; Karanam, B.; Peng, S.; Wang, J.W.; Jiang, R.; Tanno, T.; Orlowski, R.Z.; Matsui, W.; Zhao, M.; Rudek, M.A.; et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 2013, 24, 791–805. [Google Scholar]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar]
- Mazumdar, T.; Gorgun, F.M.; Sha, Y.; Tyryshkin, A.; Zeng, S.; Hartmann-Petersen, R.; Jørgensen, J.P.; Hendil, K.B.; Eissa, N.T. Regulation of NF-kappaB activity and inducible nitric oxide synthase by regulatory particle non-ATPase subunit 13 (Rpn13). Proc. Natl. Acad. Sci. USA 2010, 107, 13854–13859. [Google Scholar]
- Martin, M.; Sun, M.; Motolani, A.; Lu, T. The Pivotal Player: Components of NF-κB Pathway as Promising Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 7429. [Google Scholar]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Targeting STAT3 Signaling Pathway in Colorectal Cancer. Biomedicines 2021, 9, 1016. [Google Scholar]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar]
- Zhang, N.; Zuo, Y.; Peng, Y.; Zuo, L. Function of N6-Methyladenosine Modification in Tumors. J. Oncol. 2021, 2021, 6461552. [Google Scholar]
- Han, X.; Wang, M.; Zhao, Y.L.; Yang, Y.; Yang, Y.G. RNA methylations in human cancers. Semin. Cancer Biol. 2021, 75, 97–115. [Google Scholar]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar]
- Liu, T.; Li, C.; Jin, L.; Li, C.; Wang, L. The Prognostic Value of m6A RNA Methylation Regulators in Colon Adenocarcinoma. Med. Sci. Monit. 2019, 25, 9435–9445. [Google Scholar]
- Li, H.; Wang, C.; Lan, L.; Yan, L.; Li, W.; Evans, I.; Ruiz, E.J.; Su, Q.; Zhao, G.; Wu, W.; et al. METTL3 promotes oxaliplatin resistance of gastric cancer CD133+ stem cells by promoting PARP1 mRNA stability. Cell Mol. Life Sci. 2022, 79, 135. [Google Scholar]
- Shen, M.; Li, Y.; Wang, Y.; Shao, J.; Zhang, F.; Yin, G.; Chen, A.; Zhang, Z.; Zheng, S. N(6)-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021, 47, 102151. [Google Scholar]
- Li, Z.; Peng, Y.; Li, J.; Chen, Z.; Chen, F.; Tu, J.; Lin, S.; Wang, H. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020, 11, 2578. [Google Scholar]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene 2020, 726, 144132. [Google Scholar]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, Z.; Wu, Z.; Zhang, F.; Yang, M.; Lu, Z.; Yu, B.; Long, F.; Guo, Y.; Yang, K.; Hu, G.; et al. GMEB2 Promotes the Growth of Colorectal Cancer by Activating ADRM1 Transcription and NF-κB Signalling and Is Positively Regulated by the m6A Reader YTHDF1. Cancers 2022, 14, 6046. https://doi.org/10.3390/cancers14246046
Ning Z, Wu Z, Zhang F, Yang M, Lu Z, Yu B, Long F, Guo Y, Yang K, Hu G, et al. GMEB2 Promotes the Growth of Colorectal Cancer by Activating ADRM1 Transcription and NF-κB Signalling and Is Positively Regulated by the m6A Reader YTHDF1. Cancers. 2022; 14(24):6046. https://doi.org/10.3390/cancers14246046
Chicago/Turabian StyleNing, Zhengping, Zhiwei Wu, Fan Zhang, Ming Yang, Zhixing Lu, Bowen Yu, Fei Long, Yihang Guo, Kaiyan Yang, Gui Hu, and et al. 2022. "GMEB2 Promotes the Growth of Colorectal Cancer by Activating ADRM1 Transcription and NF-κB Signalling and Is Positively Regulated by the m6A Reader YTHDF1" Cancers 14, no. 24: 6046. https://doi.org/10.3390/cancers14246046