
An Inflammatory Response-Related Gene Signature Can Predict the Prognosis and Impact the Immune Status of Lung Adenocarcinoma
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Extraction (TCGA-LUAD and GEO(GSE31210) Cohorts)
2.2. Establishment and Verification of the Prognosis Model of IRGs
2.3. Functional Annotation
2.4. Tumor Microenvironment (TME) and Immune Response Analysis
2.5. ChemoSensitivity
2.6. RNA Isolation and qRT-PCR Analysis
2.7. Immunohistochemistry (IHC)
2.8. Statistical Analysis
3. Results
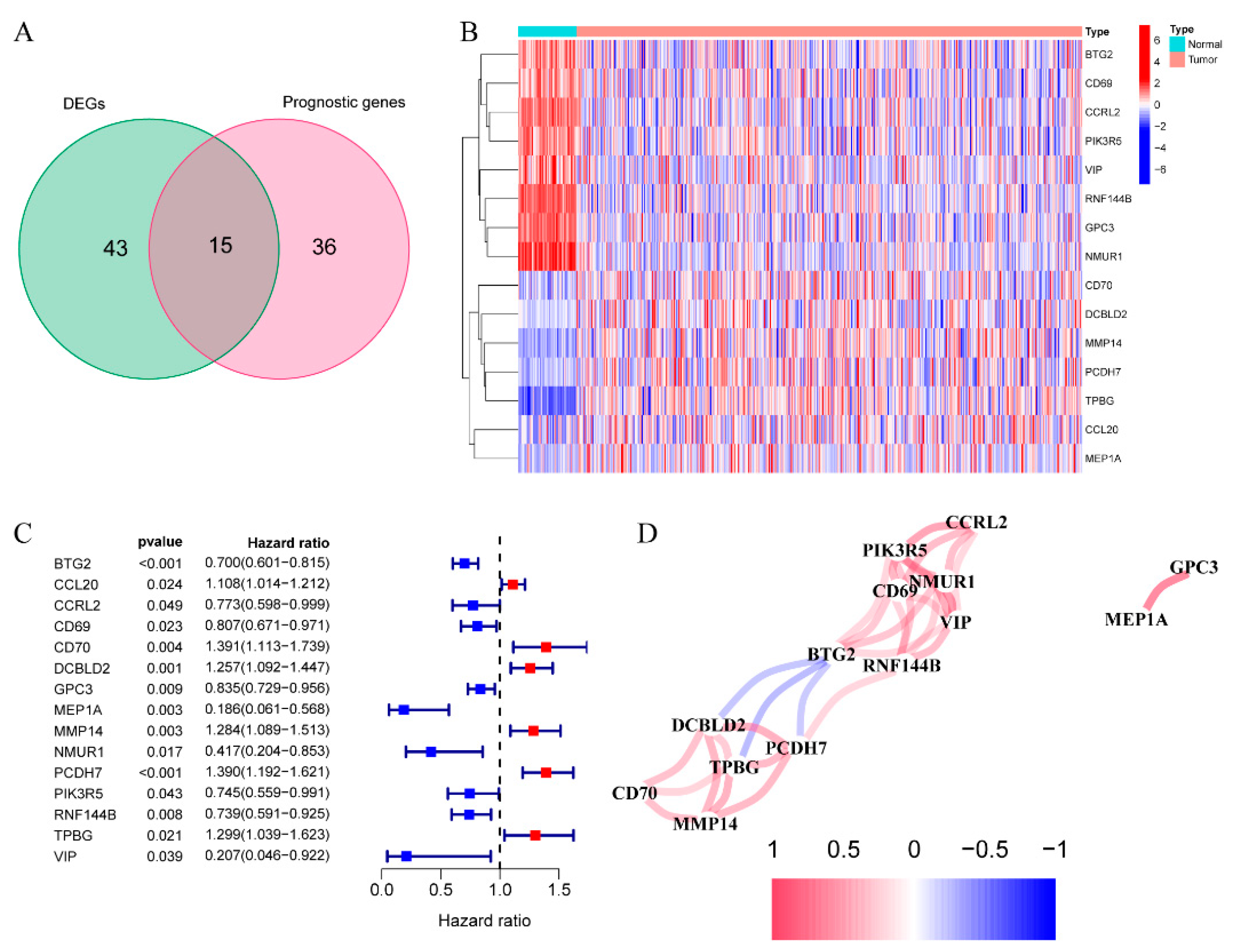
3.1. Discovery of Prognostic IRGs from TCGA Dataset
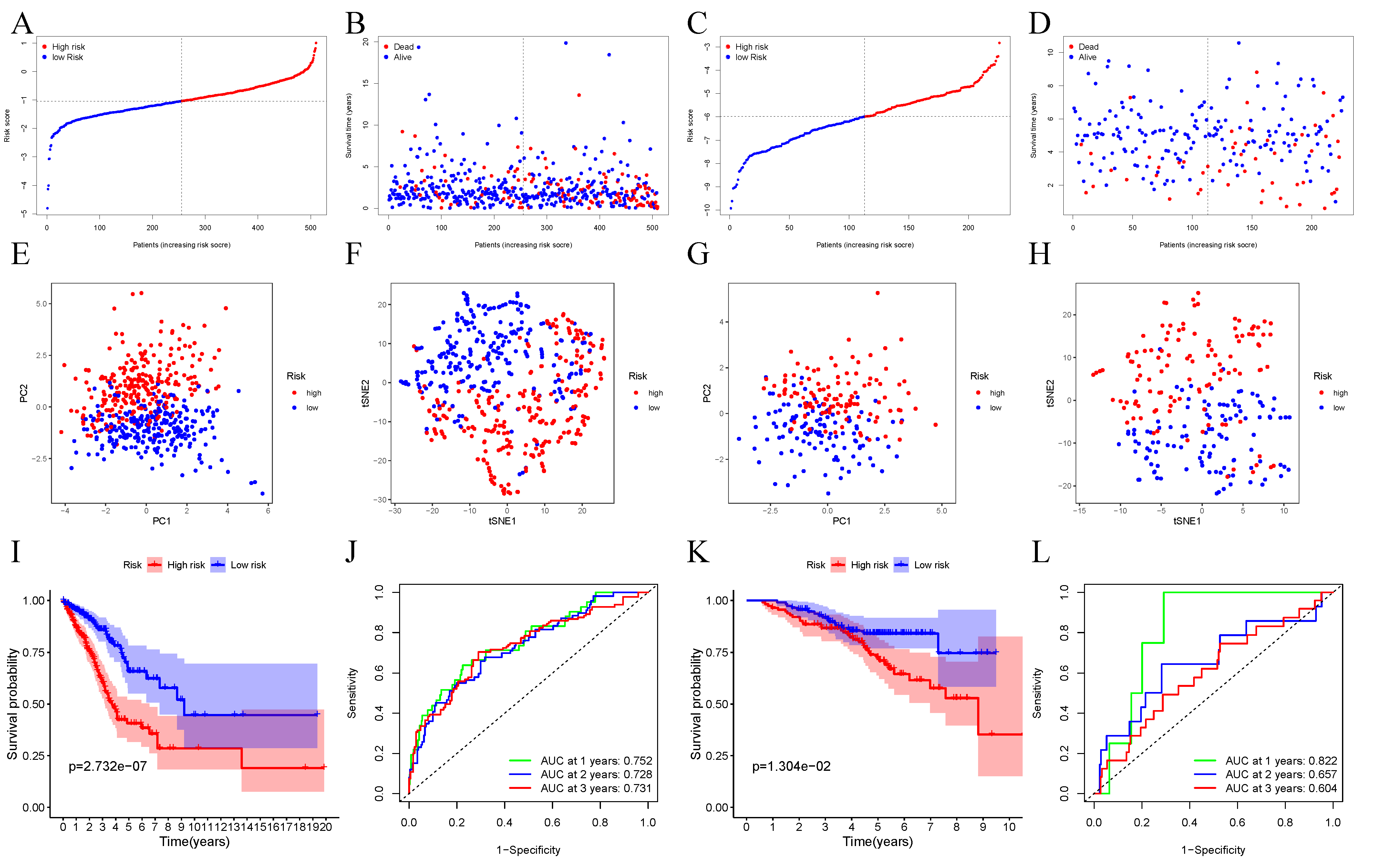
3.2. Establishment of the Prognosis Model for TCGA Dataset
3.3. Verification of 10-Gene Signature Using GEO Dataset
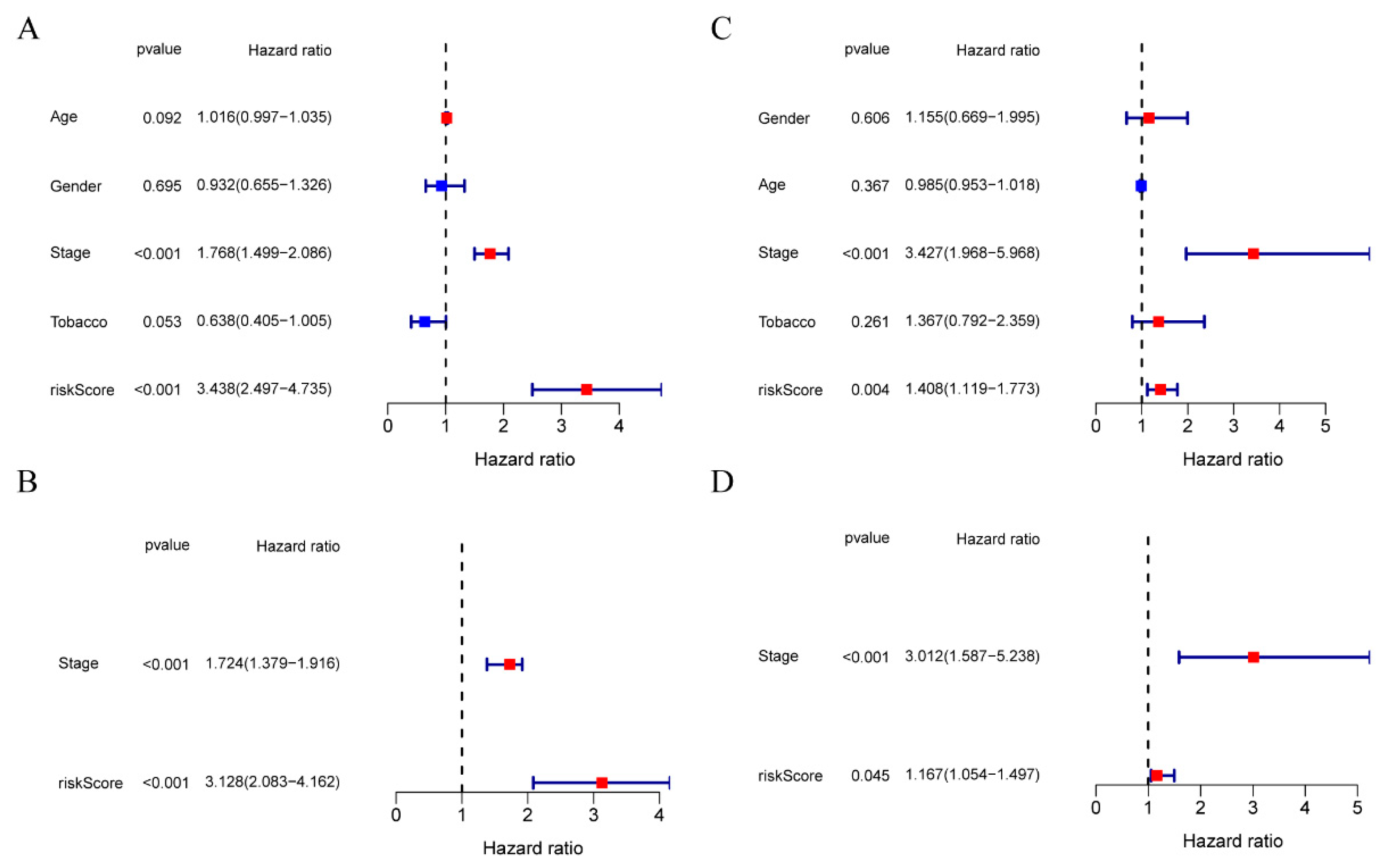
3.4. The Ability of Our 10-Gene Signature to Independently Predict Prognosis
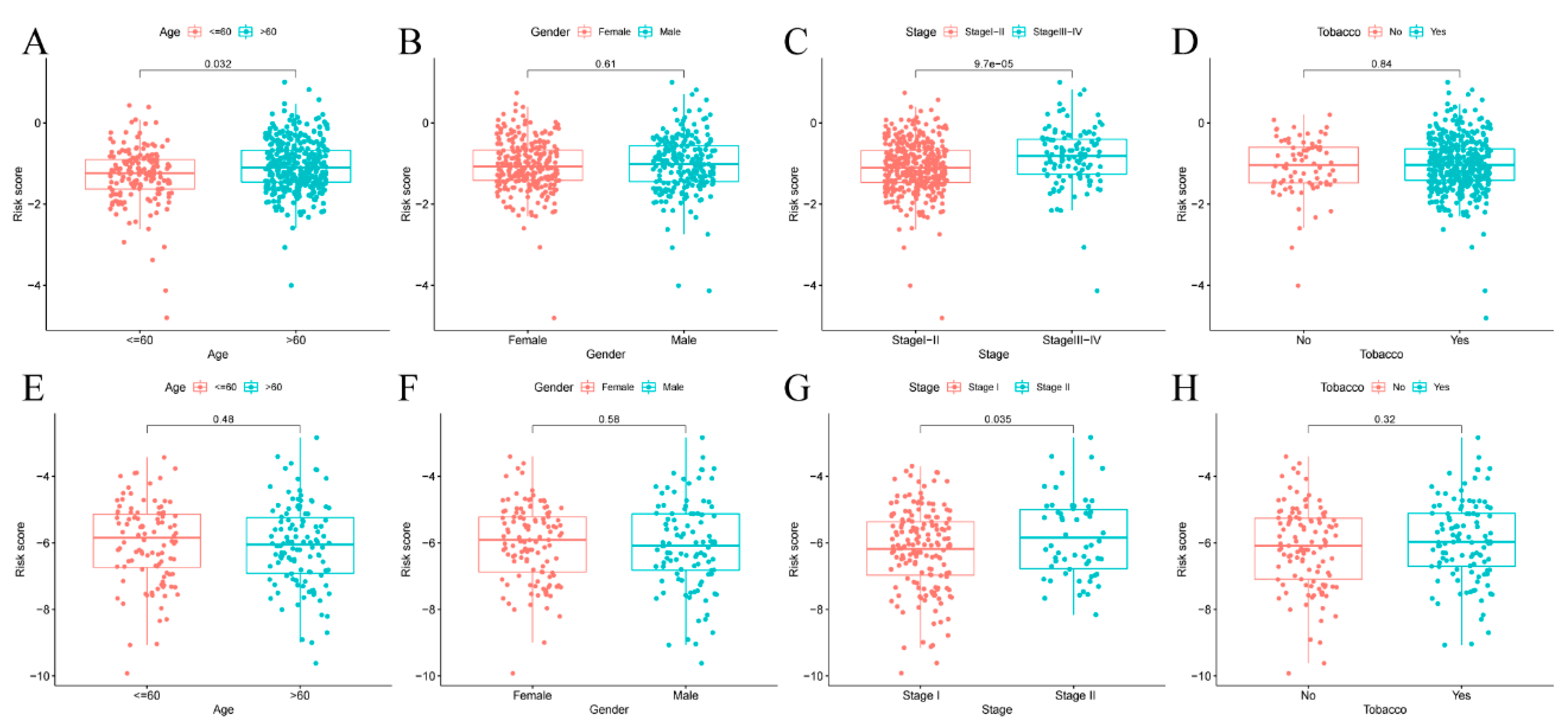
3.5. Risk Score of Prognosis Model and Clinical Characteristics
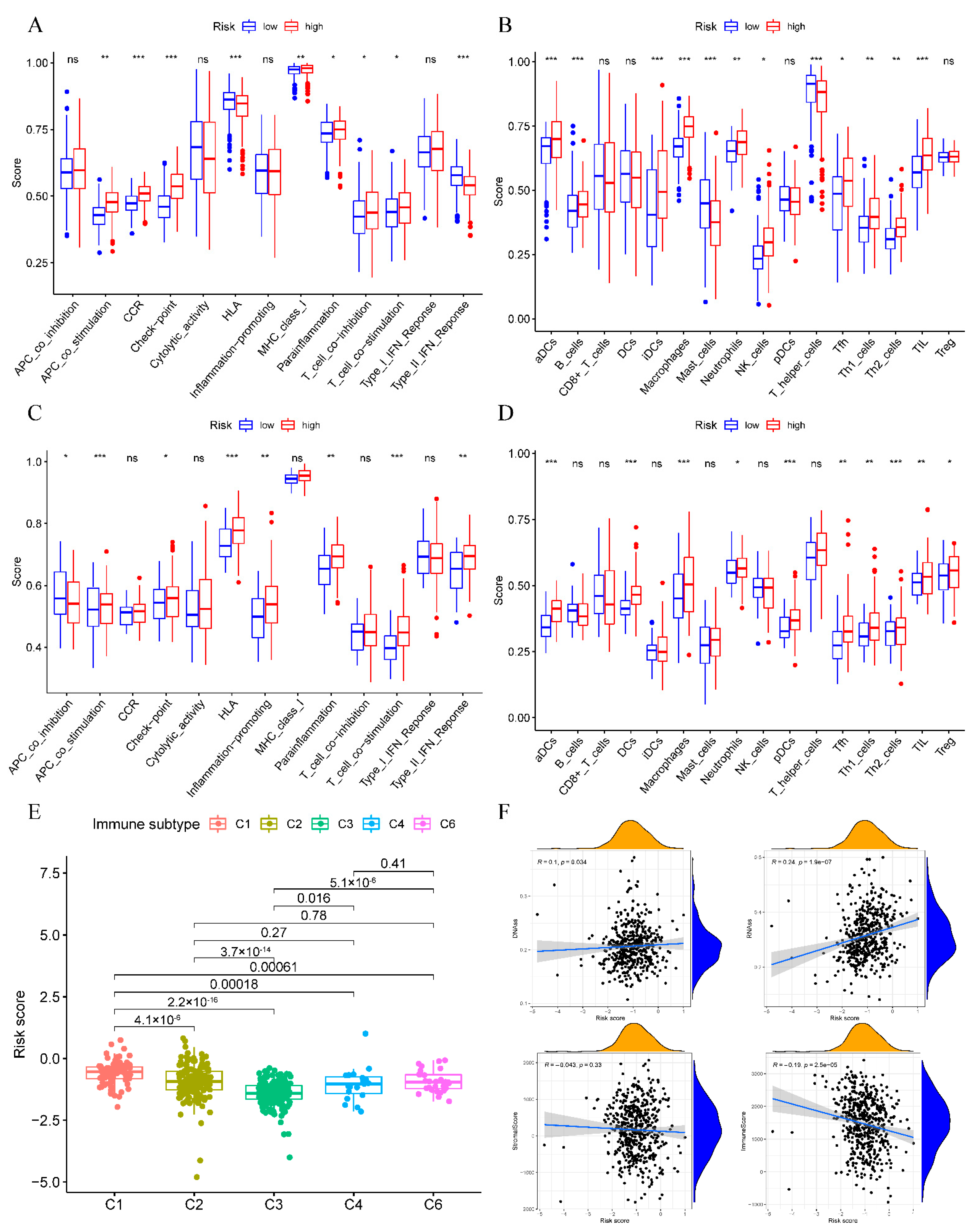
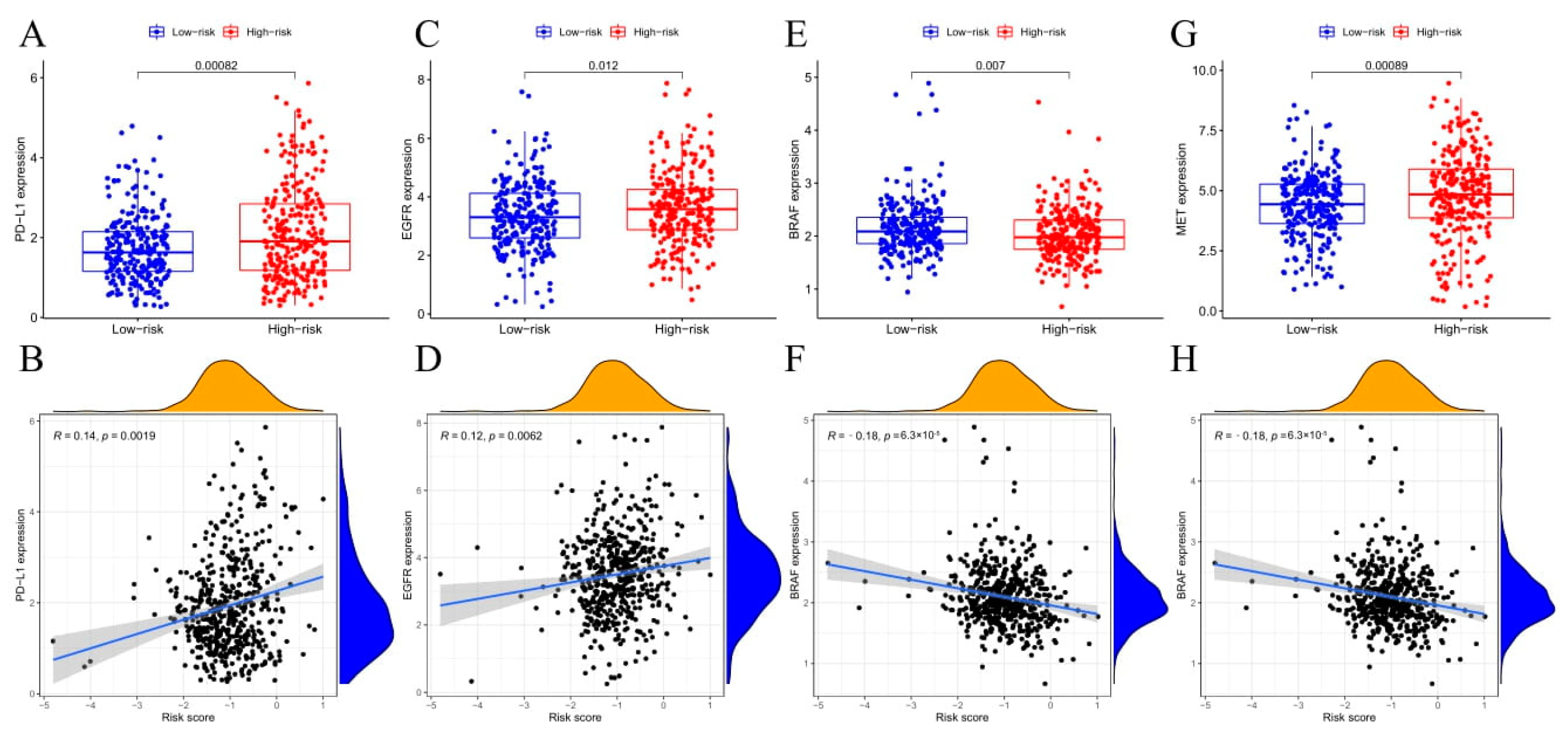
3.6. Immune Status and TME Analyses
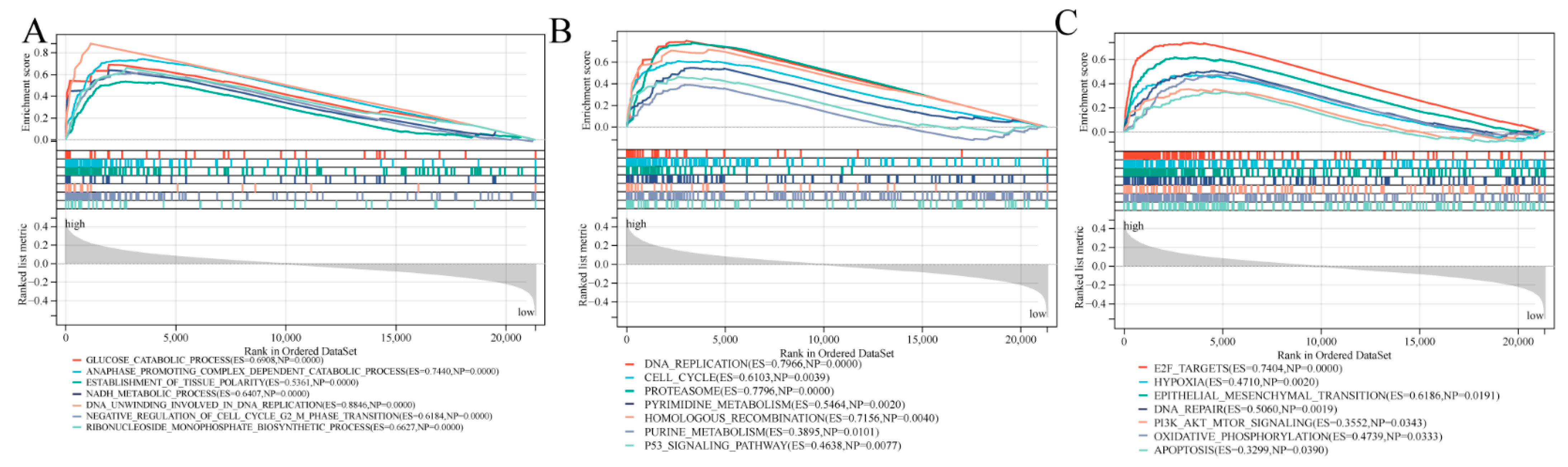
3.7. Biological Function and Pathway Analyses
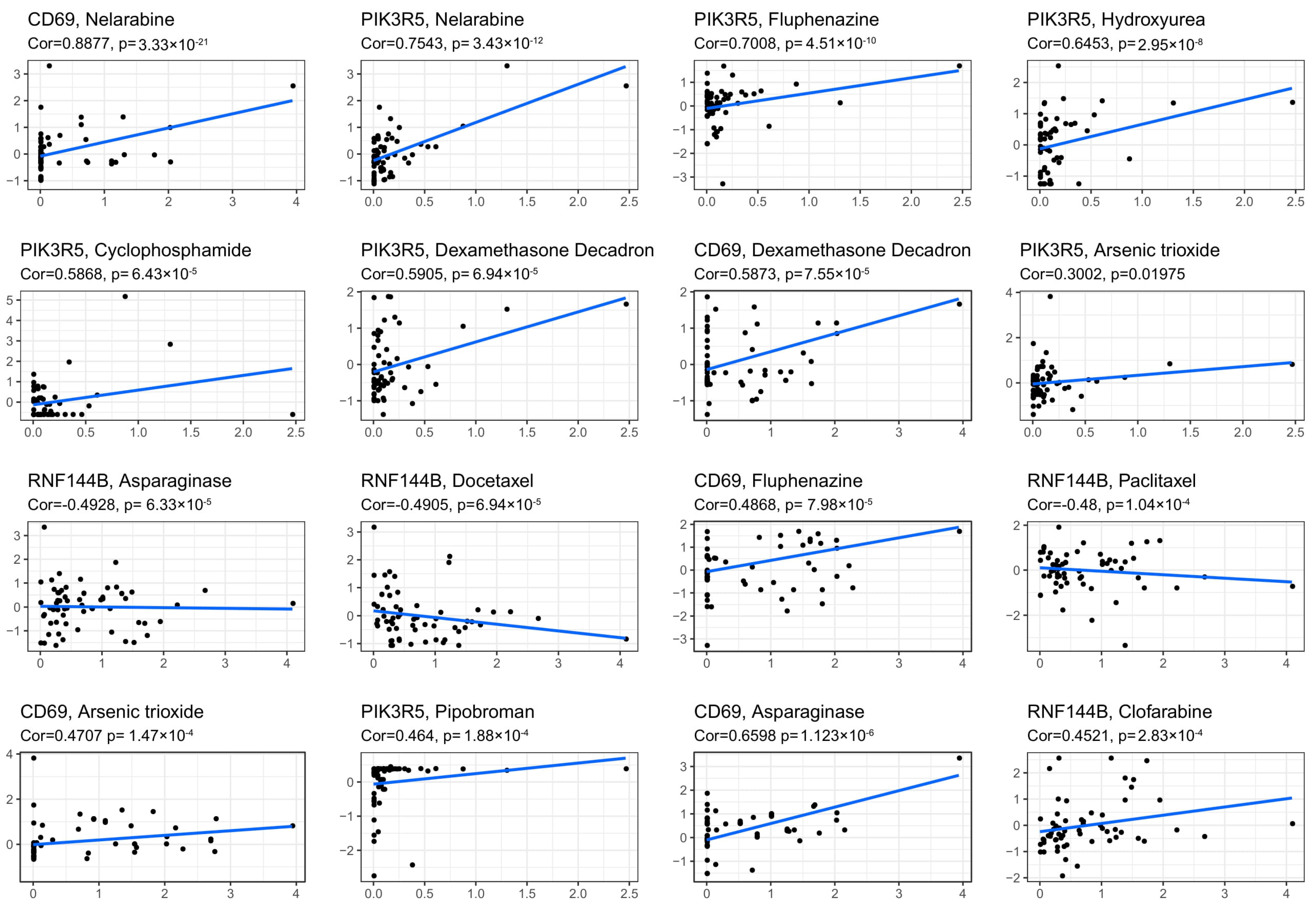
3.8. Prognostic Gene Levels and Drug Sensitivity of Cancer Cells
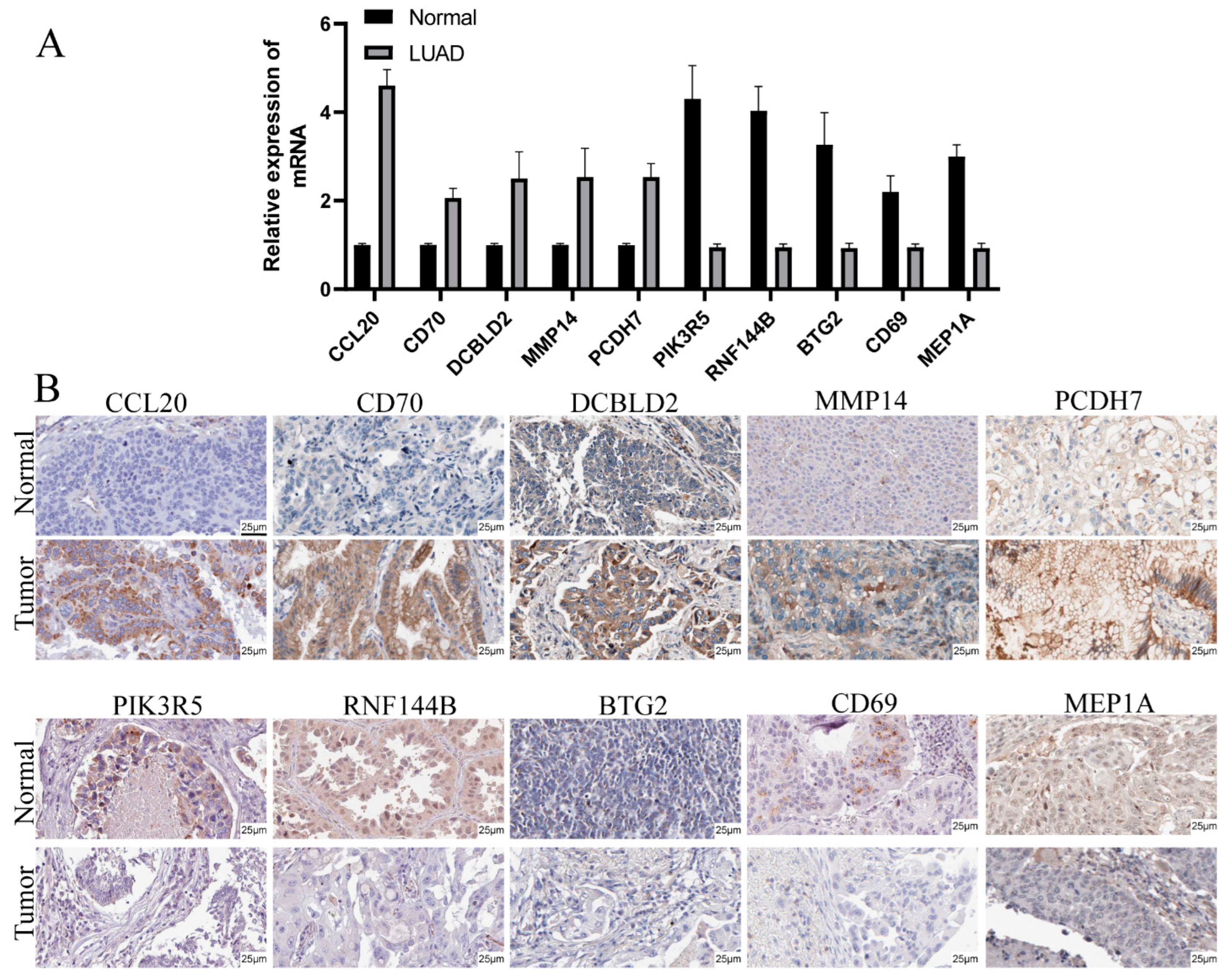
3.9. Validation of mRNA and Protein Expression of Prognostic Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.C. The systemic inflammation-based Glasgow Prognostic Score: A decade of experience in patients with cancer. Cancer Treat. Rev. 2013, 39, 534–540. [Google Scholar] [CrossRef]
- Moik, F.; Zöchbauer-Müller, S.; Posch, F.; Pabinger, I.; Ay, C. Systemic Inflammation and Activation of Haemostasis Predict Poor Prognosis and Response to Chemotherapy in Patients with Advanced Lung Cancer. Cancers 2020, 12, 1619. [Google Scholar] [CrossRef]
- Lin, Z.; Xu, Q.; Miao, D.; Yu, F. An Inflammatory Response-Related Gene Signature Can Impact the Immune Status and Predict the Prognosis of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 644416. [Google Scholar] [CrossRef]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Seijo, L.M.; Peled, N.; Ajona, D.; Boeri, M.; Field, J.K.; Sozzi, G.; Pio, R.; Zulueta, J.J.; Spira, A.; Massion, P.P.; et al. Biomarkers in Lung Cancer Screening: Achievements, Promises, and Challenges. J. Thorac. Oncol. 2019, 14, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maly, V.; Maly, O.; Kolostova, K.; Bobek, V. Circulating Tumor Cells in Diagnosis and Treatment of Lung Cancer. In Vivo 2019, 33, 1027–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadowska, K.; Bil-Lula, I.; Trembecki, Ł.; Śliwińska-Mossoń, M. Genetic Markers in Lung Cancer Diagnosis: A Review. Int. J. Mol. Sci. 2020, 21, 4569. [Google Scholar] [CrossRef]
- Song, C.; Pan, S.; Li, D.; Hao, B.; Lu, Z.; Lai, K.; Li, N.; Geng, Q. Comprehensive analysis reveals the potential value of inflammatory response genes in the prognosis, immunity, and drug sensitivity of lung adenocarcinoma. BMC Med. Genom. 2022, 15, 198. [Google Scholar] [CrossRef]
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Luo, S.; Wu, K. Immune signature-based risk stratification and prediction of immune checkpoint inhibitor’s efficacy for lung adenocarcinoma. Cancer Immunol. Immunother. 2021, 70, 1705–1719. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, T.; Zhao, D.; Qi, X.; Bao, X.; Shi, R.; Su, C. Development and validation of a hypoxia-related gene signature to predict overall survival in early-stage lung adenocarcinoma patients. Ther. Adv. Med. Oncol. 2020, 12, 1758835920937904. [Google Scholar] [CrossRef]
- Zhang, A.; Yang, J.; Ma, C.; Li, F.; Luo, H. Development and Validation of a Robust Ferroptosis-Related Prognostic Signature in Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 616271. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhang, Y.; Wang, J.; Wang, H.; Dong, N.; Su, X.; Xu, M.; Wang, X. Variations of chromosome 2 gene expressions among patients with lung cancer or non-cancer. Cell Biol. Toxicol. 2016, 32, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10. [Google Scholar] [CrossRef]
- Chen, X.; Lv, Y.; Xu, K.; Wang, X.; Zhao, Y.; Li, J.; Qin, X.; Shi, Y.; Wang, L.; Chang, A.; et al. DCBLD2 Mediates Epithelial-Mesenchymal Transition-Induced Metastasis by Cisplatin in Lung Adenocarcinoma. Cancers 2021, 13, 1403. [Google Scholar] [CrossRef]
- Du, J.; Xu, Y.; Sasada, S.; Oo, A.; Hassan, G.; Mahmud, H.; Khayrani, A.C.; Alam, J.; Kumon, K.; Uesaki, R.; et al. Signaling Inhibitors Accelerate the Conversion of mouse iPS Cells into Cancer Stem Cells in the Tumor Microenvironment. Sci. Rep. 2020, 10, 9955. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Eldakhakhny, S.; Conforti, F.; Crosbie, E.J.; Melino, G.; Sayan, B.S. Pir2/Rnf144b is a potential endometrial cancer biomarker that promotes cell proliferation. Cell Death Dis. 2018, 9, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Hao, C.; Li, X.; Zhao, H.; Chen, J.; Zhou, Q. Effects of BTG2 on proliferation inhibition and anti-invasion in human lung cancer cells. Tumour Biol. 2012, 33, 1223–1230. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, F.; Zhang, L.; Du, L.; Yan, X. Predictive value of MEP1A in cancer prognosis: A protocol for systematic review and meta-analysis. Medicine 2020, 99, e23120. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef] [Green Version]
- Shinchi, Y.; Komohara, Y.; Yonemitsu, K.; Sato, K.; Ohnishi, K.; Saito, Y.; Fujiwara, Y.; Mori, T.; Shiraishi, K.; Ikeda, K.; et al. Accurate expression of PD-L1/L2 in lung adenocarcinoma cells: A retrospective study by double immunohistochemistry. Cancer Sci. 2019, 110, 2711–2721. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J. Interferon Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma--molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA-LUAD | GSE31210 | |
|---|---|---|
| Cases | 522 | 226 |
| Age (Median, range) | 65 (33–88) | 59 (30–76) |
| Gender | ||
| Female | 280 (53.64%) | 121 (53.54%) |
| Male | 242 (43.36%) | 105 (46.46%) |
| Tabacco | ||
| Yes | 443 (84.86%) | 111 (49.12%) |
| No | 79 (15.14%) | 115 (50.88%) |
| Stage | ||
| I | 282 (54.02%) | 168 (74.34%) |
| II | 127 (24.33%) | 58 (25.66%) |
| III | 87 (16.67%) | NA |
| IV | 26 (4.98%) | NA |
| Survival Status | ||
| Alive | 384 (73.56%) | 174 (77%) |
| Dead | 138 (26.44) | 52 (23%) |
| Characteristics | TCGA-LUAD Cohort | GSE31210 Cohort | ||||
|---|---|---|---|---|---|---|
| High Risk | Low Risk | p Value | High Risk | Low Risk | p Value | |
| Age | 0.008 | 0.287 | ||||
| ≤60 year | 95 | 67 | 58 | 50 | ||
| >60 year | 160 | 188 | 55 | 63 | ||
| Gender | 0.477 | 0.23 | ||||
| Female | 134 | 142 | 65 | 56 | ||
| Male | 121 | 113 | 48 | 57 | ||
| Stage | <0.0001 | 0.034 | ||||
| I | 117 | 158 | 71 | 85 | ||
| II | 65 | 59 | 35 | 28 | ||
| III | 62 | 24 | NA | NA | ||
| IV | 11 | 14 | NA | NA | ||
| Tabacco | 1 | 0.506 | ||||
| Yes | 216 | 216 | 58 | 53 | ||
| No | 39 | 39 | 55 | 60 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Zhao, Y.; Wang, Y. An Inflammatory Response-Related Gene Signature Can Predict the Prognosis and Impact the Immune Status of Lung Adenocarcinoma. Cancers 2022, 14, 5744. https://doi.org/10.3390/cancers14235744
Shi Y, Zhao Y, Wang Y. An Inflammatory Response-Related Gene Signature Can Predict the Prognosis and Impact the Immune Status of Lung Adenocarcinoma. Cancers. 2022; 14(23):5744. https://doi.org/10.3390/cancers14235744
Chicago/Turabian StyleShi, Yubo, Yingchun Zhao, and Yuanyong Wang. 2022. "An Inflammatory Response-Related Gene Signature Can Predict the Prognosis and Impact the Immune Status of Lung Adenocarcinoma" Cancers 14, no. 23: 5744. https://doi.org/10.3390/cancers14235744