
Targeting Menin and CD47 to Address Unmet Needs in Acute Myeloid Leukemia

Abstract
:Simple Summary
Abstract
1. Introduction: Continued Need for New Therapies
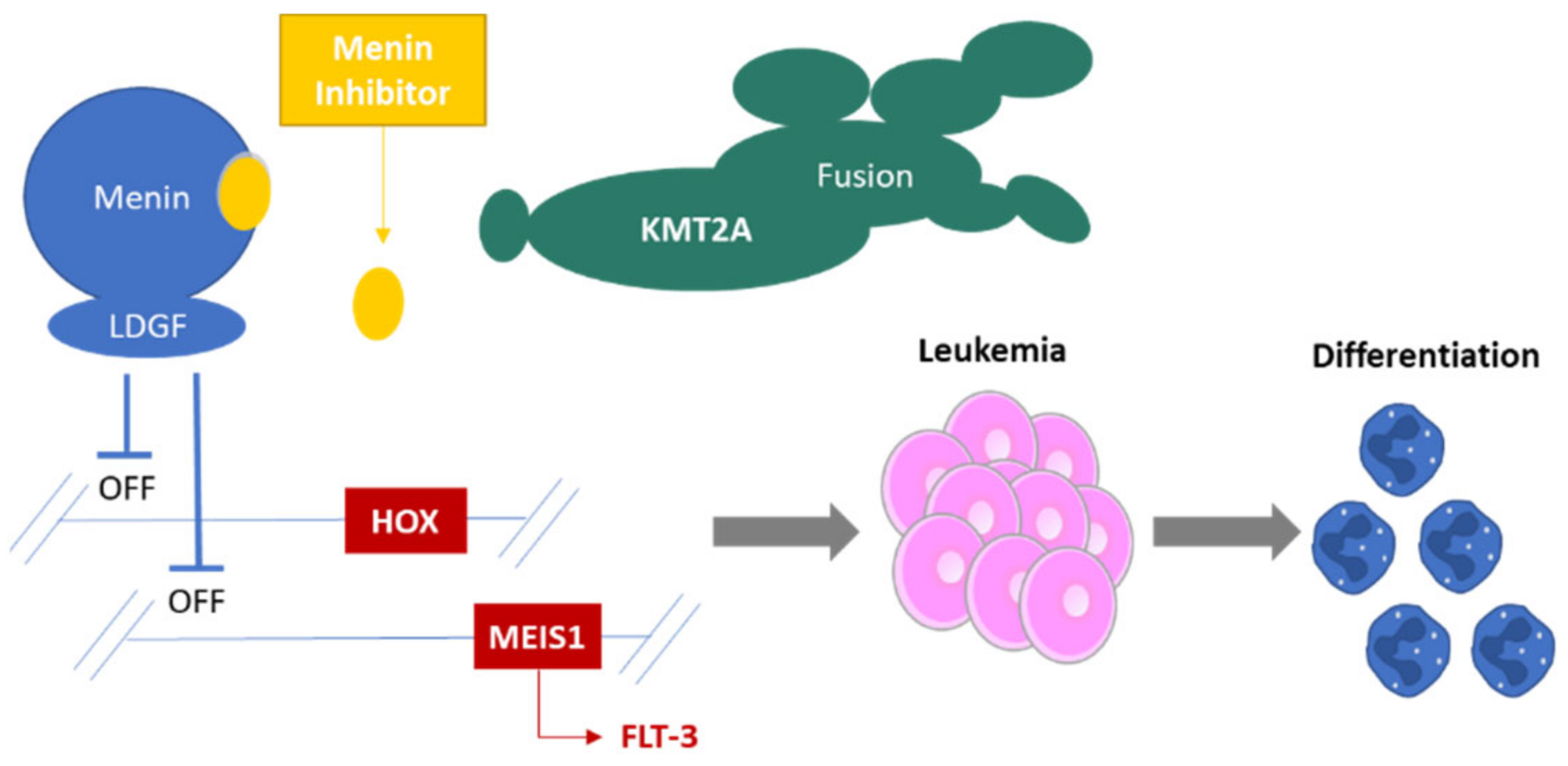
2. Menin Biology: A Short Introduction
3. Menin Inhibition
4. Targeting Menin in Acute Leukemia
5. Future Steps in Development
6. Anti-CD47 Antibodies
7. Targeting CD47 in Acute Myeloid Leukemia
8. Future Development
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Song, Y.; Wu, K. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: Estimates based on the global burden of disease study 2017. J. Hematol. Oncol. 2020, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Bewersdorf, J.P.; Stahl, M.F.; Halene, S.; Zeidan, A.M. Are we moving the needle for patients with TP53-mutated acute myeloid leukemia? Cancers 2022, 14, 2434. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Uckelmann, H.J.; Armstrong, S.A. Chromatin complexes maintain self-renewal of myeloid progenitors in AML: Opportunities for therapeutic intervention. Stem Cell Rep. 2020, 15, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Szczepański, T.; Harrison, C.J.; van Dongen, J.J.M. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol. 2010, 11, 880–889. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, L.; Nomdedéu, J.F.; Villamor, N.; Guardia, R.; Colomer, D.; Ribera, J.M.; Torres, J.P.; Berlanga, J.J.; Fernández, C.; Llorente, A.; et al. Acute myeloid leukemia with MLL rearrangements: Clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia 2003, 17, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Thiede, C.; Koch, S.; Creutzig, E.; Steudel, C.; Illmer, T.; Schaich, M.; Ehninger, G.; Dsil, F.T.D.S.L. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 2006, 107, 4011–4020. [Google Scholar] [CrossRef]
- Liu, Y.; He, P.; Liu, F.; Shi, L.; Zhu, H.; Zhao, J.; Wang, Y.; Cheng, X.; Zhang, M. Prognostic significance of NPM1 mutations in acute myeloid leukemia: A meta-analysis. Mol. Clin. Oncol. 2014, 2, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Scacheri, P.C.; Davis, S.; Odom, D.T.; Crawford, G.E.; Perkins, S.; Halawi, M.J.; Agarwal, S.K.; Marx, S.J.; Spiegel, A.M.; Meltzer, P.S.; et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLOS Genet. 2006, 2, e51. [Google Scholar] [CrossRef] [Green Version]
- Dreijerink, K.M.; Höppener, J.W.; Timmers, H.M.; Lips, C.J. Mechanisms of disease: Multiple endocrine neoplasia type 1-relation to chromatin modifications and transcription regulation. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 562–570. [Google Scholar] [CrossRef]
- Marx, S.J.; Agarwal, S.K.; Kester, M.B.; Heppner, C.; Kim, Y.S.; Skarulis, M.C.; James, L.A.; Goldsmith, P.K.; Saggar, S.K.; Park, S.Y.; et al. Multiple endocrine neoplasia type 1: Clinical and genetic features of the hereditary endocrine neoplasias. Recent Prog. Horm. Res. 1999, 54, 397–438. [Google Scholar]
- Yokoyama, A.; Somervaille, T.C.P.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Mbangkollo, D.; Burnett, R.; McCabe, N.; Thirman, M.; Gill, H.; Yu, H.; Rowley, J.D.; Diaz, M.O. The human MLL gene: Nucleotide sequence, homology to the Drosophila trx zinc-finger domain, and alternative splicing. DNA Cell Biol. 1995, 14, 475–483. [Google Scholar] [CrossRef]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.D. The critical role of chromosome translocations in human leukemias. Annu. Rev. Genet. 1998, 32, 495–519. [Google Scholar] [CrossRef]
- Jude, C.D.; Climer, L.; Xu, D.; Artinger, E.; Fisher, J.K.; Ernst, P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 2007, 1, 324–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, P.; Fisher, J.K.; Avery, W.; Wade, S.; Foy, D.; Korsmeyer, S.J. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell 2004, 6, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, R.A.; Pettengell, R.; Pandha, H.S.; Morgan, R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013, 27, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, H.; Deguchi, K.; Aono, A.; Tani, Y.; Kishimoto, T.; Komori, T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 1998, 92, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.L.; Yu, B.D.; Li, B.; Hanson, R.; Korsmeyer, S.J. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 1997, 90, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Aplan, P.D. Chromosomal translocations involving the MLL gene: Molecular mechanisms. DNA Repair 2006, 5, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.E.; Milne, T.A.; Bloyer, S.; Galoian, K.; Shen, W.; Gibbs, D.; Brock, H.W.; Slany, R.; Hess, J.L. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell 2003, 4, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Cozzio, A.; Passegué, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Ayton, P.M.; Cleary, M.L. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 2001, 20, 5695–5707. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.-H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef]
- Novotny, E.; Compton, S.; Liu, P.P.; Collins, F.S.; Chandrasekharappa, S.C. In vitro hematopoietic differentiation of mouse embryonic stem cells requires the tumor suppressor menin and is mediated by Hoxa9. Mech. Dev. 2009, 126, 517–522. [Google Scholar] [CrossRef]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin Associates with a trithorax family histone methyltransferase complex and with the Hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar] [CrossRef]
- Schwaller, J. Learning from mouse models of MLL fusion gene-driven acute leukemia. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194550. [Google Scholar] [CrossRef]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T. An Mll–AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Li, Q.; Hudson, W.A.; Kumar, A.; Kirchhof, N.; Kersey, J.H. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood 2006, 108, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Kohlmann, A.; Schoch, C.; Dugas, M.; Schnittger, S.; Hiddemann, W.; Kern, W.; Haferlach, T. New insights into MLL gene rearranged acute leukemias using gene expression profiling: Shared pathways, lineage commitment, and partner genes. Leukemia 2005, 19, 953–964. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; Boer, M.D.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Li, Z.; Chen, P.; Su, R.; Hu, C.; Li, Y.; Elkahloun, A.G.; Zuo, Z.; Gurbuxani, S.; Arnovitz, S.; Weng, H.; et al. PBX3 and MEIS1 cooperate in hematopoietic cells to drive acute myeloid leukemias characterized by a core transcriptome of the MLL—Rearranged DISEASE. Cancer Res. 2016, 76, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Martinez, A.-M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant transformation initiated by Mll-AF9: Gene dosage and critical target cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Brunangelo, F.; Cristina, M.; Giuseppe, S.; Francesco Lo, C.; Daniela, D.; Patrick, B.; Fabrizio, P.; Marco, M.; Maria Paola, M.; Stefano, P.; et al. NPM1 mutations and cytoplasmic nucleophosmin are mutually exclusive of recurrent genetic abnormalities: A comparative analysis of 2562 patients with acute myeloid leukemia. Haematologica 2008, 93, 439–442. [Google Scholar]
- Collins, C.T.; Hess, J.L. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin. Hematol. 2016, 23, 354–361. [Google Scholar] [CrossRef]
- Kühn, M.W.M.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.-W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [Green Version]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell 2019, 36, 660–673. [Google Scholar] [CrossRef]
- Numata, M.; Shiroishi, M.; Yoshikawa, K.; Haginoya, N.; Hirata, T.; Takata, Y.; Nagase, R.; Takashima, K.; Kurimoto, A.; Tanzawa, F.; et al. Development and characterization of a novel orally bioavailable menin-MLL inhibitor for treatment of acute leukemia patients with MLL-rearrangement or NPM1 mutation. AACR Annu. Meet. 2021, 2021, 1132. [Google Scholar]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [Green Version]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [Green Version]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Grembecka, J.; Belcher, A.M.; Hartley, T.; Cierpicki, T. Molecular basis of the mixed lineage leukemia-menin interaction: Implications for targeting mixed lineage leukemias. J. Biol. Chem. 2010, 285, 40690–40698. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Altman, J.K.; Issa, G.C.; Pettit, K.; DeBotten, S.; Walter, R.; Fenaux, P.; Ades, L.; Burrows, F.; Tomkinson, B.; et al. Phase 1/2 First in Human Study of the Menin-KMT2A (MLL) Inhibitor KO-539 in Patients with Relapsed or Refractory Acute Myeloid Leukemia; European Hematology Association: The Hague, The Netherlands, 2021; Abstract PB1408. [Google Scholar]
- Wang, E.; Altman, J.K.; Pettit, K.; DeBotten, S.; Walter, R.; Fenaux, P.; Burrows, F.; Tomkinson, B.; Martell, B.; Fathi, A. Preliminary data on a phase 1/2A first in human study of the menin-KMT2A (MLL) inhibitor KO-539 in patients with relapsed or refractory acute myeloid leukemia. In Proceedings of the 62nd Annual American Society of Hematology Annual Meeting and Exposition, Virtual Conference, 5–8 December 2020. [Google Scholar]
- Syndax Pharmaceuticals. Syndax Announces Positive Interim Data Demonstrating Robust Clinical Activity in Phase 1 Portion of the AUGMENT-101 Trial of SNDX-5613 in Patients with Genetically-Defined Acute Leukemias; Syndax Pharmaceuticals, Inc.: Waltham, MA, USA, 2021. [Google Scholar]
- Stein, E.M.; Aldoss, I.; DiPersio, J.F.; Stone, R.M.; Arellano, M.L.; Rosen, G.; Meyers, M.L.; Huang, Y.; Smith, S.; Bagley, R.G.; et al. Safety and efficacy of menin inhibition in patients (Pts) with MLL-rearranged and NPM1 mutant acute leukemia: A phase (Ph) 1, first-in-human study of SNDX-5613 (AUGMENT 101). Blood 2021, 138, 699. [Google Scholar] [CrossRef]
- Wang, G.G.; Pasillas, M.P.; Kamps, M.P. Meis1 programs transcription of FLT3 and cancer stem cell character, using a mechanism that requires interaction with Pbx and a novel function of the Meis1 C-terminus. Blood 2005, 106, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schönfeld, J.; Kunz, K.; Taubert, M.C.; McGeehan, G.M.; Chen, C.-W.; Mupo, A.; et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef]
- Miao, H.; Kim, E.; Chen, D.; Purohit, T.; Kempinska, K.; Ropa, J.; Klossowski, S.; Trotman, W.E.; Danet-Desnoyers, G.-A.; Cierpicki, T.; et al. Combinatorial treatment with menin and FLT3 inhibitors induces complete remission in AML models with activating FLT3 mutations. Blood 2020, 136, 2958. [Google Scholar] [CrossRef]
- Fiskus, W.; Boettcher, S.; Daver, N.; Mill, C.P.; Sasaki, K.; Birdwell, C.E.; Davis, J.A.; Takahashi, K.; Kadia, T.M.; DiNardo, C.D.; et al. Effective Menin inhibitor-based combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J. 2022, 12, 5. [Google Scholar] [CrossRef]
- Brzezinka, K.; Nevedomskaya, E.; Lesche, R.; Steckel, M.; Eheim, A.L.; Haegebarth, A.; Stresemann, C. Functional diversity of inhibitors tackling the differentiation blockage of MLL-rearranged leukemia. J. Hematol. Oncol. 2019, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Ströbel, P.; Wachter, A.; et al. Hoxa9 and Meis1 Cooperatively Induce Addiction to Syk Signaling by Suppressing miR-146a in Acute Myeloid Leukemia. Cancer Cell 2017, 31, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.R.; Byrd, J.C.; Blachly, J.S.; Bhatnagar, B.; Mims, A.S.; Orwick, S.; Lin, T.L.; Crosswell, H.E.; Zhang, D.; Minden, M.D.; et al. Entospletinib in combination with induction chemotherapy in previously untreated acute myeloid leukemia: Response and predictive significance of HOXA9 and MEIS1 expression. Clin. Cancer Res. 2020, 26, 5852–5859. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Structure, function and inhibition of critical protein-protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 2021, 14, 56. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Daver, N.G.; Montalban-Bravo, G.; Jabbour, E.J.; DiNardo, C.D.; Kornblau, S.M.; Bose, P.; Alvarado, Y.; Ohanian, M.; Borthakur, G.; et al. A phase II study evaluating the combination of nivolumab (Nivo) or ipilimumab (Ipi) with azacitidine in Pts with previously treated or untreated myelodysplastic syndromes (MDS). Blood 2016, 128, 344. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Zeidan, A.M. Randomized trials with checkpoint inhibitors in acute myeloid leukaemia and myelodysplastic syndromes: What have we learned so far and where are we heading? Best Pract. Res. Clin. Haematol. 2020, 33, 101222. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.D.L.F.; et al. Immunotherapy in acute myeloid leukemia: Where we stand. Front. Oncol. 2021, 11, 656218. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Brown, E.; Hooper, L.; Ho, T.; Gresham, H. Integrin-associated protein: A 50-kD plasma membrane antigen physically and functionally associated with integrins. J. Cell Biol. 1990, 111, 2785–2794. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Khandelwal, S.; Van Rooijen, N.; Saxena, R.K. Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion 2007, 47, 1725–1732. [Google Scholar] [CrossRef]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.-P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Z.; Guo, S.; Zhang, L.; Sharma, A.; Robertson, G.P.; Huang, L. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 2013, 21, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Chung, H.; Banan, B.; Manning, P.T.; Ott, K.C.; Lin, S.; Capoccia, B.J.; Subramanian, V.; Hiebsch, R.R.; Upadhya, G.A.; et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015, 360, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Wang, J.; Willingham, S.B.; Martin, R.; Wernig, G.; Weissman, I.L. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012, 26, 2538–2545. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Tang, C.; Pachynski, R.K.; Chin, R.; Majeti, R.; Weissman, I.L. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 2011, 118, 4890–4901. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.-X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.-P.; Levin, A.M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhong, M.C.; Guo, H.; Davidson, D.; Mishel, S.; Lu, Y.; Rhee, I.; Pérez-Quintero, L.-A.; Zhang, S.; Cruz-Munoz, M.-E.; et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 2017, 544, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Boasman, K.; Simmonds, M.; Rinaldi, C. CALR and CD47: An insight into their roles in the disease progression of MDS and MPN. J. Blood Disord. Transfus. 2018, 10, 1–5. [Google Scholar] [CrossRef]
- Uno, S.; Kinoshita, Y.; Azuma, Y.; Tsunenari, T.; Yoshimura, Y.; Iida, S.; Kikuchi, Y.; Yamada-Okabe, H.; Fukushima, N. Antitumor activity of a monoclonal antibody against CD47 in xenograft models of human leukemia. Oncol. Rep. 2007, 17, 1189–1194. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood J. Am. Soc. Hematol. 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 2020, 30, 481–496. [Google Scholar] [CrossRef]
- Wang, B.; Lim, C.B.; Yan, J.; Li, L.; Wang, J.; Little, J.B.; Yuan, Z.-M. MDMX phosphorylation-dependent p53 downregulation contributes to an immunosuppressive tumor microenvironment. J. Mol. Cell Biol. 2020, 12, 713–722. [Google Scholar] [CrossRef]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.H.Y.; Chai, V.; Lee, V.; Dodge, K.; Truong, T.; Wong, M.; Johnson, L.; Linderoth, E.; Pang, X.; Winston, J.; et al. TTI-621 (SIRPαFc), a CD47-blocking cancer immunotherapeutic, triggers phagocytosis of lymphoma cells by multiple polarized macrophage subsets. PLoS ONE 2017, 12, e0187262. [Google Scholar] [CrossRef] [Green Version]
- Johnson, Z.; Papaioannou, A.; Bernard, L.; Cosimo, E.; Daubeuf, B.; Richard, F.; Chauchet, X.; Moine, V.; Broyer, L.; Shang, L.; et al. Bispecific antibody targeting of CD47/CD19 to promote enhanced phagocytosis of patient B lymphoma cells. J. Clin. Oncol. 2015, 33 (Suppl. 15), e14016. [Google Scholar] [CrossRef]
- Sallman, D.A.; Donnellan, W.; Asch, A.; Lee, D.; Al Malki, M.; Pollyea, D.; Kambhampati, S.; Komrokji, R.S.; Van Elk, J.; Lin, M.; et al. The First-in-Class Anti-CD47 Antibody HU5F9-G4 is Active and Well Tolerated Alone or in Combination with Azacitidine in AML and MDS Patients; Initial Phase 1B Results; European Hematology Association: The Hague, The Netherlands, 2019; Volume Abstract: S878. [Google Scholar]
- Vyas, P.; Knapper, S.; Kelly, R.; Salim, R.; Lubowiecki, M.; Royston, D.; Johnson, H.; Roberts, C.; Chen, J.Y.; Agoram, B.; et al. Initial Phase 1 Results of the First-In-Class Anti-CD47 Antibody HU5F9-G4 in Relapsed/Refractory Acute Myeloid Leukemia Patients; European Hematology Association: The Hague, The Netherlands, 2018; Volume PF232. [Google Scholar]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.M.; Seet, C.S.; Tallman, M.S.; Wei, X.; Li, Y.F.; Hock, R.N.; Burgess, M.R.; Hege, K.; et al. A phase I study of CC-90002, a monoclonal antibody targeting CD47, in patients with relapsed and/or refractory (R/R) Acute Myeloid Leukemia (AML) and high-risk myelodysplastic syndromes (MDS): Final results. Blood 2019, 134 (Suppl. 1), 1320. [Google Scholar] [CrossRef]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Malki, M.M.A.; Larson, R.A.; Asch, A.S.; Mannis, G.N.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in frontline TP53m AML patients: Phase 1b results. J. Clin. Oncol. 2022, 40 (Suppl. 16), 7020. [Google Scholar] [CrossRef]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.; Murphy, M. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; McKenna, K.M.; Choi, T.S.; Duan, M.J.; Brown, M.L.; Stewart, J.J.; Sompalli, M.K.; Vyas, P.; Schrier, S.; Majeti, R.; et al. RBC-specific CD47 pruning confers protection and underlies the transient anemia in patients treated with anti-CD47 antibody 5F9. Blood 2018, 132, 2327. [Google Scholar] [CrossRef]
- Boasman, K.; Bridle, C.; Simmonds, M.; Rinaldi, C. Role of pro-phagocytic calreticulin and anti-phagocytic CD47 in MDS and MPN models treated with azacytidine or ruxolitinib. In Haematolgica; Ferrata Storti Foundation: Pavia, Italy, 2017; p. 763. [Google Scholar]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Valentin, R.; Peluso, M.O.; Lehmberg, T.Z.; Adam, A.; Zhang, L.; Armet, C.M.; Guerriero, J.L.; Lee, B.H.; Palombella, V.J.; Holland, P.M.; et al. The fully human anti-CD47 antibody SRF231 has dual-mechanism antitumor activity against chronic lymphocytic leukemia (CLL) cells and increases the activity of both rituximab and venetoclax. Blood 2018, 132, 4393. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Molecule | Phase | Enrollment | Status | Regimen | NCT | Population |
|---|---|---|---|---|---|---|
| SNDX-5613 | I/II | 186 | Recruiting | Monotherapy | NCT04065399 | Relapsed/refractory (R/R) MPAL, KMT2Ar AML, NPM1m AML |
| KO-539 | I/II | 100 | Recruiting | Monotherapy | NCT04067336 | Relapsed/refractory all patients expansion: KMT2Ar and NPM1m R/R AML [57] |
| JNJ-75276617 | I | 110 | Recruiting | Monotherapy | NCT04811560 | Relapsed/refractory: KMT2Ar, NPM1m AML |
| BMF-219 | I/II | 100 | Recruiting | Monotherapy | NCT05153330 | Relapsed/refractory all patients (with and without strong CYP3A4 inhibitors) |
| DS-1594b | I/II | 122 | Recruiting | Combination (ven/aza or Mini-HCV) | NCT04752163 | Monotherapy in relapsed/Refractory KMT2Ar AML Monotherapy in relapsed/Refractory NPM1m AML Aza/Ven Combo in R/R NPM1 or KMT2Ar AML Mini-HCVD in R/R ALL with KMT2Ar |
| Agent | Phase | Enrollment | Status | Regimen | NCT | Population |
|---|---|---|---|---|---|---|
| Magrolimab (Hu5F9-G4) | III | 520 | Recruiting | Combination with aza | NCT04313881 | Treatment naïve (TN) higher risk MDS |
| Magrolimab (Hu5F9-G4) | III | 346 | Recruiting | Combination with aza | NCT04778397 | TP53 mutated TN unfit (TNU) AML; TP53 mutated TN fit AML |
| Magrolimab (Hu5F9-G4) | Ib/II | 98 | Recruiting | Combination with ven/aza | NCT04435691 | Ib: Relapsed/Refractory (R/R) AML; II: TNU AML or TN < 75 years old with high risk cytogenetics +/− TP53 |
| Magrolimab (Hu5F9-G4) | Ib | 287 | Recruiting [105] | Combination with aza | NCT03248479 | R/R AML, TNU AML, RBC transfusion dependent low risk MDS |
| Magrolimab (Hu5F9-G4) | Ib | 13 | Completed | Combination with Atezolizumab | NCT03922477 | R/R AML |
| Magrolimab (Hu5F9-G4) | Ia | 20 | Completed [106] | Monotherapy | NCT02678338 | R/R AML; R/R higher risk MDS |
| TJ011133 (TJC4, Lemzoparlimab) | IIA | 80 | Recruiting | Combination with aza | NCT04202003 | TNU AML or TN higher risk MDS |
| TJ011133 (TJC4, Lemzoparlimab) | Ib | 120 | Recruiting | Combination with aza or ven/aza | NCT04912063 | TNU AML; high risk MDS |
| ALX148 (Evorpacept *) | I/II | 97 | Recruiting | Combination with ven/aza | NCT04755244 | R/R AML or new AML in patients ineligible for standard induction |
| AK117 | Ib/II | 160 | Recruiting | Combination with aza | NCT04980885 | AML, newly diagnosed or R/R |
| DSP107 * | Ib/II | 36 | Recruiting | Combination with aza | NCT04937166 | TNU AML, TN MDS, R/R MDS/CMML |
| TTI-621 | Ia/Ib | 260 | Recruiting | Monotherapy | NCT02663518 | R/R AML |
| TTI-662 | Ia/Ib | 150 | Recruiting | Ia: single agent 1b: combination with ven/aza | NCT03530683 | TN TP53-mutated AML aza+TTI-662; TNU AML TTI-662 + aza/ven |
| IBI188 | Ia | 12 | Recruiting | Combination with aza | NCT04485065 | TN higher risk MDS |
| IBI188 | Ib | 126 | Recruiting | Combination with aza | NCT04485052 | R/R AML, TNU AML |
| CC-90002 | I | 28 | Terminated [107] | Monotherapy | NCT02641002 | R/R AML or high risk MDS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthews, A.H.; Pratz, K.W.; Carroll, M.P. Targeting Menin and CD47 to Address Unmet Needs in Acute Myeloid Leukemia. Cancers 2022, 14, 5906. https://doi.org/10.3390/cancers14235906
Matthews AH, Pratz KW, Carroll MP. Targeting Menin and CD47 to Address Unmet Needs in Acute Myeloid Leukemia. Cancers. 2022; 14(23):5906. https://doi.org/10.3390/cancers14235906
Chicago/Turabian StyleMatthews, Andrew H., Keith W. Pratz, and Martin P. Carroll. 2022. "Targeting Menin and CD47 to Address Unmet Needs in Acute Myeloid Leukemia" Cancers 14, no. 23: 5906. https://doi.org/10.3390/cancers14235906