
The Study of the Extracellular Matrix in Chronic Inflammation: A Way to Prevent Cancer Initiation?
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. ECM as More Than a Physical Support: ECM Major Components, Properties and Functions
1.2. ECM-Cells Interplay
1.3. ECM Deposition
1.4. Degradation of ECM and MMPs Activity
1.5. ECM Dynamics under Pathological Conditions
2. Role of ECM in IME
2.1. The Immune Microenvironment and ECM Components Interactions
2.2. ECM Damaging and Remodeling during Chronic Inflammation
2.3. Key Immune Cells Driving ECM Remodeling during Chronic Inflammation
2.4. ECM-Derived Fragments as Modulators of Chemotactic Activity for Immune Cells
2.5. Alterations in ECM Components Affect Immune Cells’ Activity
2.6. ECM-Fragments Influence Gene Expression of Inflammatory Cells
3. From Chronic Inflammation to Cancer: The Main Correlations
3.1. Main Inflammatory Pathways Correlated with Tumorigenesis and Colitis-Associated Cancer
3.2. ECM Deposition in TME
3.3. Tumorigenic Alterations in ECM Composition
MMPs’ Role in Shaping TME
3.4. Tumorigenic ECM and Its Remodeling Influence on Immune Cells within Tumor Mass
4. Pre-Clinical Models’ Application in TME and IME
4.1. Scaffold-Free and Scaffold-Based Systems
4.2. Patient-Derived Scaffold Obtained by Tissue Decellularization
4.3. 3D Patient-Derived dECM Models: Novel Strategies to Study IME to Prevent TME?
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2D | (two-dimensional) |
| 3D | (three-dimensional) |
| ADAMs | (disintegrin and metalloproteinases) |
| ADAMTS | (thrombospondin motifs) |
| ASKVKV | (Ala-Ser-Lys-Val-Lys-Val) |
| BFGF | (fibroblast growth factor) |
| BM | (basement membranes) |
| BMP | (bone morphogenetic protein) |
| CXCL | (C-X-C motif ligand) |
| CAC | (colitis-associated colon cancer) |
| CAFs | (cancer-associated fibroblasts) |
| COMP | (cartilage oligomeric matrix protein) |
| COX-2 | (cyclooxygenase 2) |
| CRC | (colorectal cancer) |
| CRLM | (colorectal liver metastasis) |
| DAMPs | (damage-associated molecular patterns) |
| DDR | (DNA damage response) |
| DDrs | (discoidin domain receptors) |
| dECM | (decellularized ECM) |
| DEFA3 | (Defensin Alpha 3) |
| EAC | (esophageal adenocarcinoma) |
| EBP | (elastin binding protein) |
| ECM | (extracellular matrix) |
| ED-A and ED-B | (extra domain A-B) |
| EGF | (epidermal growth factor) |
| EHS | (Engelbreth-Holm-Swarm) c |
| EMT | (epithelial to mesenchymal transition) |
| EP | (E-type prostanoid) |
| ERK | (extracellular-regulated kinase) |
| FAK | (focal adhesion kinase) |
| FGF-2 | (fibroblast growth factor) |
| FMLP | (formyl met-leu-phe) |
| FOLFIRI | (folinic acid, fluorouracil acid and irinotecan) |
| FOLFIRINOX | (folinic acid, fluoruracil acid, irinotecan and oxaliplatin) |
| GAGs | (glycosaminoglycans) |
| GLUT | (glucose transporters) |
| GM-CSF | (Granulocyte-Macrophage Colony-Stimulating Factor) |
| HA | (hyaluronic acid) |
| HIF | (hypoxia-inducible factor) |
| HC | (Healhty colon) |
| HL | (Healthy Liver) |
| IBD | (inflammatory bowel disease) |
| IFNγ | (interferon gamma) |
| ILs | (interleukins) |
| IM | (interstitial matrix) |
| IME | (inflammatory microenvironment) |
| JNK | (c-Jun N-terminal kinase) |
| LAP | (latency-associated protein) |
| LTBP | (Latent TGFβ binding protein) |
| LAP | (latency-associated protein) |
| LOX | (lysyl oxidases) |
| LOXLs | (LOX-like proteins) |
| MAPK | (mitogen-activated protein kinase) |
| MCs | (mast cells) |
| MT1-MMP | (membrane-type 1 matrix metalloproteinase) |
| MHs | (microporous hydrogels) |
| MIP | (macrophage inflammatory protein) |
| MMPs | (metalloproteinases) |
| MHC | (Major Histocompatibility Complex) |
| MTT | (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) |
| MYC | (myelocytomatosis) |
| NETs | (neutrophil extracellular traps) |
| NF-kB | (nuclear factor kappa-light-chain-enhancer of activated B cells) |
| NK | (natural killer) |
| OPN | (Osteopontin) |
| PAI | (plasminogen activator inhibitor) |
| PAMPs | (pathogen-associated patterns) |
| PDAC | (pancreatic ductal adenocarcinoma) |
| PDGF | (platelet-derived growth factor) |
| PDGF-EC | (platelet derived endothelial cell growth factor) |
| PGP | (Pro-Gly-Pro) |
| PGs | (proteoglycans) |
| PPARs | (peroxisome proliferator-activated receptors) |
| PGE2 | (Prostaglandin E2) |
| PTEN | (Phosphatase and tensin homolog) |
| PTPs | (protein tyrosine phosphatases) |
| PVDF | (polyvinylidene fluoride) |
| RA | (rheumatoid arthritis) |
| RNI | (reactive nitrogen intermediate) |
| RNS | (reactive nitrogen species) |
| ROCK1 | (Rho-associated protein kinase 1) |
| ROS | (reactive oxygen species) |
| SDF | (stromal cell-derived factor 1) |
| SERPINE2 | (serine proteinase inhibitor-E2) |
| SIKVAV | (Ser-Ile-Lys-Val-Ala-Val) |
| SRC | (Proto-oncogene tyrosine-protein kinase) |
| TAMs | (Tumor associated macrophages) |
| TANs | (tumor-associated neutrophils) |
| TGF-β | (transforming growth factor β) |
| Th17 | (T-helper IL-17 producing cells) |
| TINKs | (tumor-infiltrating natural killer cells) |
| TIMP | (tissue inhibitor of metalloproteinases) |
| TLRs | (Toll-like receptors) |
| TME | (tumor microenvironment) |
| TNF-α | (tumor necrosis factor α) |
| TNFR | (tumor necrosis factor receptor superfamily) |
| Tregs | (Regulatory T cells) |
| TSPs | (Thrombospondins) |
| VEGF | (vascular endothelial growth factors) |
References
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues That Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, S.S.; Weinberg, R.A. The Tumour-Induced Systemic Environment as a Critical Regulator of Cancer Progression and Metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-Associated Stromal Cells as Key Contributors to the Tumor Microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.A.; Niland, S. The Extracellular Matrix in Tumor Progression and Metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Yue, B. Biology of the Extracellular Matrix: An Overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, T.B.; Seim, E. Association between Invasiveness, Inflammatory Reaction, Desmoplasia and Survival in Colorectal Cancer. J. Clin. Pathol. 1989, 42, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Kai, F.; Laklai, H.; Weaver, V.M. Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [Green Version]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Decellularized Extracellular Matrix for Bioengineering Physiomimetic 3D in Vitro Tumor Models. Trends Biotechnol. 2020, 38, 1397–1414. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Durães, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized Human Colorectal Cancer Matrices Polarize Macrophages towards an Anti-Inflammatory Phenotype Promoting Cancer Cell Invasion via CCL18. Biomaterials 2017, 124, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Sensi, F.; D’Angelo, E.; Piccoli, M.; Pavan, P.; Mastrotto, F.; Caliceti, P.; Biccari, A.; Corallo, D.; Urbani, L.; Fassan, M.; et al. Recellularized Colorectal Cancer Patient-Derived Scaffolds as in Vitro Pre-Clinical 3D Model for Drug Screening. Cancers 2020, 12, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Fishbein, A.; Wang, W.; Yang, H.; Yang, J.; Hallisey, V.M.; Deng, J.; Verheul, S.M.L.; Hwang, S.H.; Gartung, A.; Wang, Y.; et al. Resolution of Eicosanoid/Cytokine Storm Prevents Carcinogen and Inflammation-Initiated Hepatocellular Cancer Progression. Proc. Natl. Acad. Sci. USA 2020, 117, 21576–21587. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular Matrix Assembly: A Multiscale Deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr. Top. Dev. Biol. 2018, 130, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.P.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin Regulates Latent Transforming Growth Factor-Beta (TGF Beta) by Controlling Matrix Assembly of Latent TGF Beta-Binding Protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brézillon, S.; Götte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef]
- Norton, W.H.J.; Ledin, J.; Grandel, H.; Neumann, C.J. HSPG Synthesis by Zebrafish Ext2 and Extl3 Is Required for Fgf10 Signalling during Limb Development. Development 2005, 132, 4963–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N.; et al. An Organoid-Based Preclinical Model of Human Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef] [Green Version]
- Lokmic, Z.; Lämmermann, T.; Sixt, M.; Cardell, S.; Hallmann, R.; Sorokin, L. The Extracellular Matrix of the Spleen as a Potential Organizer of Immune Cell Compartments. Semin. Immunol. 2008, 20, 4–13. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix in Development and Morphogenesis: A Dynamic View. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular Matrix Structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in Wound Healing, Fibrosis and Tumor Stroma: High Potential Targets for Therapeutics and Drug Delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Vachon, P.H. Integrin Signaling, Cell Survival, and Anoikis: Distinctions, Differences, and Differentiation. J. Signal Transduct. 2011, 2011, 738137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; DuFort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The Cancer Glycocalyx Mechanically Primes Integrin-Mediated Growth and Survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afratis, N.A.; Nikitovic, D.; Multhaupt, H.A.B.; Theocharis, A.D.; Couchman, J.R.; Karamanos, N.K. Syndecans—Key Regulators of Cell Signaling and Biological Functions. FEBS J. 2017, 284, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-Mediated Mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional Homeostasis and the Malignant Phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.I.; Kang, I.; You, W.-K.; McDonald, D.M.; Weaver, V.M. In Situ Force Mapping of Mammary Gland Transformation. Integr. Biol. 2011, 3, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Kölsch, V.; Seher, T.; Fernandez-Ballester, G.J.; Serrano, L.; Leptin, M. Control of Drosophila Gastrulation by Apical Localization of Adherens Junctions and RhoGEF2. Science 2007, 315, 384–386. [Google Scholar] [CrossRef]
- Montell, D.J. Morphogenetic Cell Movements: Diversity from Modular Mechanical Properties. Science 2008, 322, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Ciobanasu, C.; Faivre, B.; Le Clainche, C. Actomyosin-Dependent Formation of the Mechanosensitive Talin-Vinculin Complex Reinforces Actin Anchoring. Nat. Commun. 2014, 5, 3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.Y.-F.; Chan, M.K.-K.; Li, J.S.-F.; Chan, A.S.-W.; Tang, P.C.-T.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef] [PubMed]
- Barbazán, J.; Matic Vignjevic, D. Cancer Associated Fibroblasts: Is the Force the Path to the Dark Side? Curr. Opin. Cell Biol. 2019, 56, 71–79. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile Extracellular Regulators in Cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef]
- Remillard, T.C.; Bratslavsky, G.; Jensen-Taubman, S.; Stetler-Stevenson, W.G.; Bourboulia, D. Molecular Mechanisms of Tissue Inhibitor of Metalloproteinase 2 in the Tumor Microenvironment. Mol. Cell. Ther. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Niland, S.; Eble, J.A. Hold on or Cut? Integrin- and MMP-Mediated Cell-Matrix Interactions in the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 22, 238. [Google Scholar] [CrossRef]
- Taniwaki, K.; Fukamachi, H.; Komori, K.; Ohtake, Y.; Nonaka, T.; Sakamoto, T.; Shiomi, T.; Okada, Y.; Itoh, T.; Itohara, S.; et al. Stroma-Derived Matrix Metalloproteinase (MMP)-2 Promotes Membrane Type 1-MMP-Dependent Tumor Growth in Mice. Cancer Res. 2007, 67, 4311–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, N.; Allen, E.; Apel, I.J.; Gyetko, M.R.; Weiss, S.J. Matrix Metalloproteinases Regulate Neovascularization by Acting as Pericellular Fibrinolysins. Cell 1998, 95, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef] [PubMed]
- Koorman, T.; Jansen, K.A.; Khalil, A.; Haughton, P.D.; Visser, D.; Rätze, M.A.K.; Haakma, W.E.; Sakalauskaitè, G.; van Diest, P.J.; de Rooij, J.; et al. Spatial Collagen Stiffening Promotes Collective Breast Cancer Cell Invasion by Reinforcing Extracellular Matrix Alignment. Oncogene 2022, 41, 2458–2469. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A Tense Situation: Forcing Tumour Progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-Mediated Cellular Tension Drives Increased Tissue Stiffness and β-Catenin Activation to Induce Epidermal Hyperplasia and Tumor Growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Petrey, A.C.; de la Motte, C.A. The Extracellular Matrix in IBD: A Dynamic Mediator of Inflammation. Curr. Opin. Gastroenterol. 2017, 33, 234–238. [Google Scholar] [CrossRef]
- Shimshoni, E.; Adir, I.; Afik, R.; Solomonov, I.; Shenoy, A.; Adler, M.; Puricelli, L.; Sabino, F.; Savickas, S.; Mouhadeb, O.; et al. Distinct Extracellular-Matrix Remodeling Events Precede Symptoms of Inflammation. Matrix Biol. 2021, 96, 47–68. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil Elastase, Proteinase 3 and Cathepsin G: Physicochemical Properties, Activity and Physiopathological Functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Shamamian, P.; Schwartz, J.D.; Pocock, B.J.; Monea, S.; Whiting, D.; Marcus, S.G.; Mignatti, P. Activation of Progelatinase A (MMP-2) by Neutrophil Elastase, Cathepsin G, and Proteinase-3: A Role for Inflammatory Cells in Tumor Invasion and Angiogenesis. J. Cell. Physiol. 2001, 189, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Delclaux, C.; Delacourt, C.; D’Ortho, M.P.; Boyer, V.; Lafuma, C.; Harf, A. Role of Gelatinase B and Elastase in Human Polymorphonuclear Neutrophil Migration across Basement Membrane. Am. J. Respir. Cell Mol. Biol. 1996, 14, 288–295. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, N.R.; Smith, A.M. Matrix Metalloproteases Role in Bowel Inflammation and Inflammatory Bowel Disease: An up to Date Review. Inflamm. Bowel Dis. 2014, 20, 2379–2393. [Google Scholar] [CrossRef]
- Derkacz, A.; Olczyk, P.; Olczyk, K.; Komosinska-Vassev, K. The Role of Extracellular Matrix Components in Inflammatory Bowel Diseases. J. Clin. Med. 2021, 10, 1122. [Google Scholar] [CrossRef]
- Low, Q.E.; Drugea, I.A.; Duffner, L.A.; Quinn, D.G.; Cook, D.N.; Rollins, B.J.; Kovacs, E.J.; DiPietro, L.A. Wound Healing in MIP-1alpha(-/-) and MCP-1(-/-) Mice. Am. J. Pathol. 2001, 159, 457–463. [Google Scholar] [CrossRef]
- Moodley, Y.; Rigby, P.; Bundell, C.; Bunt, S.; Hayashi, H.; Misso, N.; McAnulty, R.; Laurent, G.; Scaffidi, A.; Thompson, P.; et al. Macrophage Recognition and Phagocytosis of Apoptotic Fibroblasts Is Critically Dependent on Fibroblast-Derived Thrombospondin 1 and CD36. Am. J. Pathol. 2003, 162, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The Role of Macrophage Phenotype in Vascularization of Tissue Engineering Scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human Neutrophils Uniquely Release TIMP-Free MMP-9 to Provide a Potent Catalytic Stimulator of Angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Kolesnikoff, N.; Chen, C.-H.; Samuel, M.S. Interrelationships between the Extracellular Matrix and the Immune Microenvironment That Govern Epithelial Tumour Progression. Clin. Sci. 2022, 136, 361–377. [Google Scholar] [CrossRef]
- Vangansewinkel, T.; Geurts, N.; Quanten, K.; Nelissen, S.; Lemmens, S.; Geboes, L.; Dooley, D.; Vidal, P.M.; Pejler, G.; Hendrix, S. Mast Cells Promote Scar Remodeling and Functional Recovery after Spinal Cord Injury via Mouse Mast Cell Protease 6. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 2040–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, M.; Grimbaldeston, M.A.; Nakae, S.; Piliponsky, A.M.; Tsai, M.; Galli, S.J. Mast Cells in the Promotion and Limitation of Chronic Inflammation. Immunol. Rev. 2007, 217, 304–328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.O.; Lampinen, M.; Rollman, O.; Sommerhoff, C.P.; Paivandy, A.; Pejler, G. Mast Cell Chymase Affects the Functional Properties of Primary Human Airway Fibroblasts: Implications for Asthma. J. Allergy Clin. Immunol. 2022, 149, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Tchougounova, E.; Pejler, G.; Abrink, M. The Chymase, Mouse Mast Cell Protease 4, Constitutes the Major Chymotrypsin-like Activity in Peritoneum and Ear Tissue. A Role for Mouse Mast Cell Protease 4 in Thrombin Regulation and Fibronectin Turnover. J. Exp. Med. 2003, 198, 423–431. [Google Scholar] [CrossRef]
- Tchougounova, E.; Lundequist, A.; Fajardo, I.; Winberg, J.-O.; Abrink, M.; Pejler, G. A Key Role for Mast Cell Chymase in the Activation of Pro-Matrix Metalloprotease-9 and pro-Matrix Metalloprotease-2. J. Biol. Chem. 2005, 280, 9291–9296. [Google Scholar] [CrossRef] [Green Version]
- Adair-Kirk, T.L.; Senior, R.M. Fragments of Extracellular Matrix as Mediators of Inflammation. Int. J. Biochem. Cell Biol. 2008, 40, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Senior, R.M.; Hinek, A.; Griffin, G.L.; Pipoly, D.J.; Crouch, E.C.; Mecham, R.P. Neutrophils Show Chemotaxis to Type IV Collagen and Its 7S Domain and Contain a 67 KD Type IV Collagen Binding Protein with Lectin Properties. Am. J. Respir. Cell Mol. Biol. 1989, 1, 479–487. [Google Scholar] [CrossRef]
- Iwabuchi, K.; Nagaoka, I.; Someya, A.; Yamashita, T. Type IV Collagen-Binding Proteins of Neutrophils: Possible Involvement of L-Selectin in the Neutrophil Binding to Type IV Collagen. Blood 1996, 87, 365–372. [Google Scholar] [CrossRef]
- Monboisse, J.C.; Garnotel, R.; Bellon, G.; Ohno, N.; Perreau, C.; Borel, J.P.; Kefalides, N.A. The Alpha 3 Chain of Type IV Collagen Prevents Activation of Human Polymorphonuclear Leukocytes. J. Biol. Chem. 1994, 269, 25475–25482. [Google Scholar] [CrossRef]
- Xu, X.; Jackson, P.L.; Tanner, S.; Hardison, M.T.; Abdul Roda, M.; Blalock, J.E.; Gaggar, A. A Self-Propagating Matrix Metalloprotease-9 (MMP-9) Dependent Cycle of Chronic Neutrophilic Inflammation. PLoS ONE 2011, 6, e15781. [Google Scholar] [CrossRef]
- Weathington, N.M.; van Houwelingen, A.H.; Noerager, B.D.; Jackson, P.L.; Kraneveld, A.D.; Galin, F.S.; Folkerts, G.; Nijkamp, F.P.; Blalock, J.E. A Novel Peptide CXCR Ligand Derived from Extracellular Matrix Degradation during Airway Inflammation. Nat. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Jackson, P.L.; Noerager, B.D.; O’Reilly, P.J.; McQuaid, D.B.; Rowe, S.M.; Clancy, J.P.; Blalock, J.E. A Novel Proteolytic Cascade Generates an Extracellular Matrix-Derived Chemoattractant in Chronic Neutrophilic Inflammation. J. Immunol. 2008, 180, 5662–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, M. Toll-like Receptor--a Potent Driving Force behind Rheumatoid Arthritis. J. Clin. Exp. Hematop. 2011, 51, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, R.M.; Gresham, H.D.; Griffin, G.L.; Brown, E.J.; Chung, A.E. Entactin Stimulates Neutrophil Adhesion and Chemotaxis through Interactions between Its Arg-Gly-Asp (RGD) Domain and the Leukocyte Response Integrin. J. Clin. Investig. 1992, 90, 2251–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, R.M.; Griffin, G.L.; Mecham, R.P. Chemotactic Activity of Elastin-Derived Peptides. J. Clin. Investig. 1980, 66, 859–862. [Google Scholar] [CrossRef]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin Fragments Drive Disease Progression in a Murine Model of Emphysema. J. Clin. Investig. 2006, 116, 753–759. [Google Scholar] [CrossRef]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Hocking, A.M.; Shinomura, T.; McQuillan, D.J. Leucine-Rich Repeat Glycoproteins of the Extracellular Matrix. Matrix Biol. 1998, 17, 1–19. [Google Scholar] [CrossRef]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.F. 3rd The Extra Domain A of Fibronectin Activates Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.B.; Brunn, G.J.; Kodaira, Y.; Platt, J.L. Receptor-Mediated Monitoring of Tissue Well-Being via Detection of Soluble Heparan Sulfate by Toll-like Receptor 4. J. Immunol. 2002, 168, 5233–5239. [Google Scholar] [CrossRef]
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and Their Proteoglycans: Host-Associated Molecular Patterns for Initiation and Modulation of Inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan Fragments: An Information-Rich System. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C Is an Endogenous Activator of Toll-like Receptor 4 That Is Essential for Maintaining Inflammation in Arthritic Joint Disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The Matrix Component Biglycan Is Proinflammatory and Signals through Toll-like Receptors 4 and 2 in Macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef]
- Schaefer, L. Extracellular Matrix Molecules: Endogenous Danger Signals as New Drug Targets in Kidney Diseases. Curr. Opin. Pharmacol. 2010, 10, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W. Hyaluronan and Its Catabolic Products in Tissue Injury and Repair. Matrix Biol. 2002, 21, 25–29. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of Lung Injury and Repair by Toll-like Receptors and Hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar] [CrossRef]
- Hodge-Dufour, J.; Noble, P.W.; Horton, M.R.; Bao, C.; Wysoka, M.; Burdick, M.D.; Strieter, R.M.; Trinchieri, G.; Puré, E. Induction of IL-12 and Chemokines by Hyaluronan Requires Adhesion-Dependent Priming of Resident but Not Elicited Macrophages. J. Immunol. 1997, 159, 2492–2500. [Google Scholar]
- McKee, C.M.; Penno, M.B.; Cowman, M.; Burdick, M.D.; Strieter, R.M.; Bao, C.; Noble, P.W. Hyaluronan (HA) Fragments Induce Chemokine Gene Expression in Alveolar Macrophages. The Role of HA Size and CD44. J. Clin. Investig. 1996, 98, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan Activate Dendritic Cells via Toll-like Receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Horton, M.R.; Shapiro, S.; Bao, C.; Lowenstein, C.J.; Noble, P.W. Induction and Regulation of Macrophage Metalloelastase by Hyaluronan Fragments in Mouse Macrophages. J. Immunol. 1999, 162, 4171–4176. [Google Scholar] [PubMed]
- Horton, M.R.; Olman, M.A.; Bao, C.; White, K.E.; Choi, A.M.; Chin, B.Y.; Noble, P.W.; Lowenstein, C.J. Regulation of Plasminogen Activator Inhibitor-1 and Urokinase by Hyaluronan Fragments in Mouse Macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L707-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, M.R.; Burdick, M.D.; Strieter, R.M.; Bao, C.; Noble, P.W. Regulation of Hyaluronan-Induced Chemokine Gene Expression by IL-10 and IFN-Gamma in Mouse Macrophages. J. Immunol. 1998, 160, 3023–3030. [Google Scholar] [PubMed]
- Horton, M.R.; McKee, C.M.; Bao, C.; Liao, F.; Farber, J.M.; Hodge-DuFour, J.; Puré, E.; Oliver, B.L.; Wright, T.M.; Noble, P.W. Hyaluronan Fragments Synergize with Interferon-Gamma to Induce the C-X-C Chemokines Mig and Interferon-Inducible Protein-10 in Mouse Macrophages. J. Biol. Chem. 1998, 273, 35088–35094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beezhold, D.H.; Personius, C. Fibronectin Fragments Stimulate Tumor Necrosis Factor Secretion by Human Monocytes. J. Leukoc. Biol. 1992, 51, 59–64. [Google Scholar] [CrossRef]
- López-Moratalla, N.; del Mar Calonge, M.; López-Zabalza, M.J.; Pérez-Mediavilla, L.A.; Subirá, M.L.; Santiago, E. Activation of Human Lymphomononuclear Cells by Peptides Derived from Extracellular Matrix Proteins. Biochim. Biophys. Acta 1995, 1265, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Marom, B.; Rahat, M.A.; Lahat, N.; Weiss-Cerem, L.; Kinarty, A.; Bitterman, H. Native and Fragmented Fibronectin Oppositely Modulate Monocyte Secretion of MMP-9. J. Leukoc. Biol. 2007, 81, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.M.; Falcone, D.J. Role of Laminin in Matrix Induction of Macrophage Urokinase-Type Plasminogen Activator and 92-KDa Metalloproteinase Expression. J. Biol. Chem. 1997, 272, 8270–8275. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, M.L.; Kibbey, M.C.; Kleinman, H.K.; Wahl, L.M. Laminin SIKVAV Peptide Induction of Monocyte/Macrophage Prostaglandin E2 and Matrix Metalloproteinases. J. Biol. Chem. 1995, 270, 10365–10368. [Google Scholar] [CrossRef] [Green Version]
- Adair-Kirk, T.L.; Atkinson, J.J.; Kelley, D.G.; Arch, R.H.; Miner, J.H.; Senior, R.M. A Chemotactic Peptide from Laminin Alpha 5 Functions as a Regulator of Inflammatory Immune Responses via TNF Alpha-Mediated Signaling. J. Immunol. 2005, 174, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- Mydel, P.; Shipley, J.M.; Adair-Kirk, T.L.; Kelley, D.G.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M. Neutrophil Elastase Cleaves Laminin-332 (Laminin-5) Generating Peptides That Are Chemotactic for Neutrophils. J. Biol. Chem. 2008, 283, 9513–9522. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Cancer and Inflammation: An Old Intuition with Rapidly Evolving New Concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Ushijima, T. Accumulation of Genetic and Epigenetic Alterations in Normal Cells and Cancer Risk. NPJ Precis. Oncol. 2019, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.-C.L.; Coudry, R.A.; Clapper, M.L.; Zhang, X.; Williams, K.-L.; Spittle, C.S.; Li, T.; Cooper, H.S. Loss of P53 Enhances the Induction of Colitis-Associated Neoplasia by Dextran Sulfate Sodium. Carcinogenesis 2007, 28, 2375–2381. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical Causes of Cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Gronke, K.; Hernández, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 Protects Intestinal Stem Cells against Genotoxic Stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef]
- Grivennikov, S.I. Inflammation and Colorectal Cancer: Colitis-Associated Neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Rowley, D.A.; Schreiber, H. Inflammation as a Tumor Promoter in Cancer Induction. Semin. Cancer Biol. 2004, 14, 433–439. [Google Scholar] [CrossRef]
- Karin, M. Nuclear Factor-KappaB in Cancer Development and Progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gehlot, P. Inflammation and Cancer: How Friendly Is the Relationship for Cancer Patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, P.M.; Lapointe, T.K.; Beck, P.L.; Buret, A.G. Mechanisms by Which Inflammation May Increase Intestinal Cancer Risk in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2010, 16, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-KappaB Signaling Pathway, Inflammation and Colorectal Cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-KappaB Functions as a Tumour Promoter in Inflammation-Associated Cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Shawki, S.; Ashburn, J.; Signs, S.A.; Huang, E. Colon Cancer: Inflammation-Associated Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 269–287. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-Beta Suppresses Tumor Progression in Colon Cancer by Inhibition of IL-6 Trans-Signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. Involvement of IL-6 in the Pathogenesis of Inflammatory Bowel Disease and Colon Cancer. Clin. Rev. Allergy Immunol. 2005, 28, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Agoff, S.N.; Brentnall, T.A.; Crispin, D.A.; Taylor, S.L.; Raaka, S.; Haggitt, R.C.; Reed, M.W.; Afonina, I.A.; Rabinovitch, P.S.; Stevens, A.C.; et al. The Role of Cyclooxygenase 2 in Ulcerative Colitis-Associated Neoplasia. Am. J. Pathol. 2000, 157, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Dubois, R.N. Colorectal Cancer Prevention and Treatment by Inhibition of Cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, M.; Kawano, S.; DuBois, R.N. Cyclooxygenase-2 Expression in Human Colon Cancer Cells Increases Metastatic Potential. Proc. Natl. Acad. Sci. USA 1997, 94, 3336–3340. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. The Role of COX-2 in Intestinal Inflammation and Colorectal Cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, H.; Brown, J.; Daikoku, T.; Ning, W.; Shi, Q.; Richmond, A.; Strieter, R.; Dey, S.K.; DuBois, R.N. CXCL1 Induced by Prostaglandin E2 Promotes Angiogenesis in Colorectal Cancer. J. Exp. Med. 2006, 203, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, H.; Shi, Q.; Katkuri, S.; Walhi, W.; Desvergne, B.; Das, S.K.; Dey, S.K.; DuBois, R.N. Prostaglandin E(2) Promotes Colorectal Adenoma Growth via Transactivation of the Nuclear Peroxisome Proliferator-Activated Receptor Delta. Cancer Cell 2004, 6, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; DuBois, R.N. PPARδ and PGE(2) Signaling Pathways Communicate and Connect Inflammation to Colorectal Cancer. Inflamm. Cell Signal. 2014, 1. [Google Scholar] [CrossRef] [Green Version]
- Sarra, M.; Pallone, F.; Macdonald, T.T.; Monteleone, G. IL-23/IL-17 Axis in IBD. Inflamm. Bowel Dis. 2010, 16, 1808–1813. [Google Scholar] [CrossRef]
- Hyun, Y.S.; Han, D.S.; Lee, A.R.; Eun, C.S.; Youn, J.; Kim, H.-Y. Role of IL-17A in the Development of Colitis-Associated Cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Pallone, F.; Monteleone, G.; Stolfi, C. Role of T(H)17 Cytokines in the Control of Colorectal Cancer. Oncoimmunology 2013, 2, e26617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundorfean, G.; Neurath, M.F.; Mudter, J. Functional Relevance of T Helper 17 (Th17) Cells and the IL-17 Cytokine Family in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2012, 18, 180–186. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.; Powrie, F. The IL23 Axis Plays a Key Role in the Pathogenesis of IBD. Gut 2007, 56, 1333–1336. [Google Scholar] [CrossRef] [Green Version]
- Heneberg, P. Paracrine Tumor Signaling Induces Transdifferentiation of Surrounding Fibroblasts. Crit. Rev. Oncol. Hematol. 2016, 97, 303–311. [Google Scholar] [CrossRef]
- White, E.S.; Muro, A.F. Fibronectin Splice Variants: Understanding Their Multiple Roles in Health and Disease Using Engineered Mouse Models. IUBMB Life 2011, 63, 538–546. [Google Scholar] [CrossRef]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The Role of the Myofibroblast in Tumor Stroma Remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic Switch Drives the Conversion of Fibroblasts into Proinvasive Cancer-Associated Fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [Green Version]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and Functional Heterogeneity of Cancer-Associated Fibroblast within the Tumor Microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-Associated Fibroblasts Modulate Growth Factor Signaling and Extracellular Matrix Remodeling to Regulate Tumor Metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, S.; Abdellatef, S.; Al Haddad, M.; Fakhoury, I.; El-Sibai, M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between Cancer and Immune Cells: Role of Tumor-Associated Macrophages in the Tumor Microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose Tissue and Adipocytes Support Tumorigenesis and Metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The Presence of Tumor Associated Macrophages in Tumor Stroma as a Prognostic Marker for Breast Cancer Patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The Clinical Significance of Hypoxia in Human Cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D.M. A Think Tank of TINK/TANKs: Tumor-Infiltrating/Tumor-Associated Natural Killer Cells in Tumor Progression and Angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The Proangiogenic Phenotype of Natural Killer Cells in Patients with Non-Small Cell Lung Cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Hempel, H.A.; Cuka, N.S.; Kulac, I.; Barber, J.R.; Cornish, T.C.; Platz, E.A.; De Marzo, A.M.; Sfanos, K.S. Low Intratumoral Mast Cells Are Associated With a Higher Risk of Prostate Cancer Recurrence. Prostate 2017, 77, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Bohaumilitzky, L.; Huber, A.-K.; Stork, E.M.; Wengert, S.; Woelfl, F.; Boehm, H. A Trickster in Disguise: Hyaluronan’s Ambivalent Roles in the Matrix. Front. Oncol. 2017, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-H.; Ji, C.-D.; Xiao, H.-L.; Zhao, H.-B.; Cui, Y.-H.; Bian, X.-W. Reorganized Collagen in the Tumor Microenvironment of Gastric Cancer and Its Association with Prognosis. J. Cancer 2017, 8, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.-C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular Matrix Remodeling: The Common Denominator in Connective Tissue Diseases. Possibilities for Evaluation and Current Understanding of the Matrix as More than a Passive Architecture, but a Key Player in Tissue Failure. Assay Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix Density-Induced Mechanoregulation of Breast Cell Phenotype, Signaling and Gene Expression through a FAK-ERK Linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [Green Version]
- Esposito, I.; Penzel, R.; Chaib-Harrireche, M.; Barcena, U.; Bergmann, F.; Riedl, S.; Kayed, H.; Giese, N.; Kleeff, J.; Friess, H.; et al. Tenascin C and Annexin II Expression in the Process of Pancreatic Carcinogenesis. J. Pathol. 2006, 208, 673–685. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Argani, P.; Hempen, P.M.; Jones, J.; Kern, S.E. The Desmoplastic Response to Infiltrating Breast Carcinoma: Gene Expression at the Site of Primary Invasion and Implications for Comparisons between Tumor Types. Cancer Res. 2002, 62, 5351–5357. [Google Scholar] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, Z.; Marshall, J.F. The Role of Integrins in TGFβ Activation in the Tumour Stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.; Clark, R.A. Fibronectin Interaction with Growth Factors in the Context of General Ways Extracellular Matrix Molecules Regulate Growth Factor Signaling. G. Ital. Dermatol. e Venereol. Organo Uff. Soc. Ital. di Dermatol. e Sifilogr. 2018, 153, 361–374. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Marchiq, I.; Pouysségur, J. Hypoxia, Cancer Metabolism and the Therapeutic Benefit of Targeting Lactate/H(+) Symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Kim, S.; Chung, H.-T.; Pae, H.-O. Reactive Oxygen Species in the Activation of MAP Kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [CrossRef]
- Chetram, M.A.; Bethea, D.A.; Odero-Marah, V.A.; Don-Salu-Hewage, A.S.; Jones, K.J.; Hinton, C. V ROS-Mediated Activation of AKT Induces Apoptosis via PVHL in Prostate Cancer Cells. Mol. Cell. Biochem. 2013, 376, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular Matrix (ECM) Stiffness and Degradation as Cancer Drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Young, J.L.; Van Vliet, K.J.; Kamm, R.D.; Discher, D.; Janmey, P.; Spatz, J.P.; Saif, T. Cell-Extracellular Matrix Mechanobiology: Forceful Tools and Emerging Needs for Basic and Translational Research. Nano Lett. 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stroka, K.M.; Konstantopoulos, K. Physical Biology in Cancer. 4. Physical Cues Guide Tumor Cell Adhesion and Migration. Am. J. Physiol. Cell Physiol. 2014, 306, C98–C109. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Ohtsuka, T.; Okada, Y.; Kanai, Y. Stromal Metalloproteinases: Crucial Contributors to the Tumor Microenvironment. Pathol. Int. 2021, 71, 1–14. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Lin, Z.; Jiang, J.; Lu, S.; Chen, M.; Que, H.; He, X.; Que, G.; Mao, J.; Xiao, J.; et al. MMP14 Regulates Cell Migration and Invasion through Epithelial-Mesenchymal Transition in Nasopharyngeal Carcinoma. Am. J. Transl. Res. 2015, 7, 950–958. [Google Scholar] [PubMed]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP Generated Matrikines. Matrix Biol. 2015, 44–46, 122–129. [Google Scholar] [CrossRef]
- Kehlet, S.N.; Sanz-Pamplona, R.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Moreno, V. Excessive Collagen Turnover Products Are Released during Colorectal Cancer Progression and Elevated in Serum from Metastatic Colorectal Cancer Patients. Sci. Rep. 2016, 6, 30599. [Google Scholar] [CrossRef] [Green Version]
- Oudin, M.J.; Weaver, V.M. Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 189–205. [Google Scholar] [CrossRef]
- Sangaletti, S.; Chiodoni, C.; Tripodo, C.; Colombo, M.P. The Good and Bad of Targeting Cancer-Associated Extracellular Matrix. Curr. Opin. Pharmacol. 2017, 35, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Sato, T.; Terai, M.; Tamura, Y.; Alexeev, V.; Mastrangelo, M.J.; Selvan, S.R. Interleukin 10 in the Tumor Microenvironment: A Target for Anticancer Immunotherapy. Immunol. Res. 2011, 51, 170–182. [Google Scholar] [CrossRef]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevach, E.M. Mechanisms of Foxp3+ T Regulatory Cell-Mediated Suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor Matrix Remodeling and Novel Immunotherapies: The Promise of Matrix-Derived Immune Biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory Roles of Versican Proteolysis in the Myeloma Microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Van De Hey, D.R.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Dhakal, B.; Pagenkopf, A.; Mushtaq, M.U.; Cunningham, A.M.; Flietner, E.; Morrow, Z.; Papadas, A.; Hope, C.; Leith, C.; Hematti, P.; et al. Versican Proteolysis Predicts Immune Effector Infiltration and Post-Transplant Survival in Myeloma. Leuk. Lymphoma 2019, 60, 2558–2562. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-Infiltrating Neutrophils Constitute the Major in Vivo Source of Angiogenesis-Inducing MMP-9 in the Tumor Microenvironment. Neoplasia 2014, 16, 771–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, D.H.; Jürgensen, H.J.; Siersbæk, M.S.; Kuczek, D.E.; Grey Cloud, L.; Liu, S.; Behrendt, N.; Grøntved, L.; Weigert, R.; Bugge, T.H. Tumor-Associated Macrophages Derived from Circulating Inflammatory Monocytes Degrade Collagen through Cellular Uptake. Cell Rep. 2017, 21, 3662–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor Macrophages Are Pivotal Constructors of Tumor Collagenous Matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef] [Green Version]
- Bryda, E.C. The Mighty Mouse: The Impact of Rodents on Advances in Biomedical Research. Mo. Med. 2013, 110, 207–211. [Google Scholar]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in Translation: Animal Models and Clinical Trials in Cancer Treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pacheco, S.; O’Driscoll, L. Pre-Clinical In Vitro Models Used in Cancer Research: Results of a Worldwide Survey. Cancers 2021, 13, 6033. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Cultures—a Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of Clinical Drug Development Fails and How to Improve It? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Shevde, L.A.; Metge, B.J.; Mitra, A.; Xi, Y.; Ju, J.; King, J.A.; Samant, R.S. Spheroid-Forming Subpopulation of Breast Cancer Cells Demonstrates Vasculogenic Mimicry via Hsa-MiR-299-5p Regulated de Novo Expression of Osteopontin. J. Cell. Mol. Med. 2010, 14, 1693–1706. [Google Scholar] [CrossRef]
- Shaheen, S.; Ahmed, M.; Lorenzi, F.; Nateri, A.S. Spheroid-Formation (Colonosphere) Assay for in Vitro Assessment and Expansion of Stem Cells in Colon Cancer. Stem Cell Rev. Rep. 2016, 12, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lv, H.-F.; Zhao, R.; Ying, M.-F.; Samuriwo, A.T.; Zhao, Y.-Z. Recent Developments in Bio-Scaffold Materials as Delivery Strategies for Therapeutics for Endometrium Regeneration. Mater. Today Bio 2021, 11, 100101. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Hutmacher, D.W. Scaffolds in Tissue Engineering Bone and Cartilage. Biomaterials 2000, 21, 2529–2543. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Kwon, H.J.; Kim, M.C.; Bae, Y.K. CD9 Expression in Colorectal Carcinomas and Its Prognostic Significance. J. Pathol. Transl. Med. 2016, 50, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, J.F.; Silva, G.A.; Azevedo, H.S.; Malafaya, P.B.; Sousa, R.A.; Silva, S.S.; Boesel, L.F.; Oliveira, J.M.; Santos, T.C.; Marques, A.P.; et al. Natural Origin Biodegradable Systems in Tissue Engineering and Regenerative Medicine: Present Status and Some Moving Trends. J. R. Soc. Interface 2007, 4, 999–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdeldin, S.; Enany, S.; Yoshida, Y.; Xu, B.; Zhang, Y.; Zureena, Z.; Lokamani, I.; Yaoita, E.; Yamamoto, T. Basics and Recent Advances of Two Dimensional- Polyacrylamide Gel Electrophoresis. Clin. Proteom. 2014, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Vila-Porcile, E.; Picart, R.; Vigny, M.; Tixier-Vidal, A.; Tougard, C. Immunolocalization of Laminin, Heparan-Sulfate Proteoglycan, Entactin, and Type IV Collagen in the Rat Anterior Pituitary. I. An in Vivo Study. Anat. Rec. 1992, 232, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic Alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.S.B.; Ponnamma, D.; Choudhary, R.; Sadasivuni, K.K. A Comparative Review of Natural and Synthetic Biopolymer Composite Scaffolds. Polymers 2021, 13, 1105. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, A.; Yang, Y. Decellularization Strategies for Regenerative Medicine: From Processing Techniques to Applications. Biomed Res. Int. 2017, 2017, 9831534. [Google Scholar] [CrossRef] [Green Version]
- Marques-Magalhães, Â.; Cruz, T.; Costa, Â.M.; Estêvão, D.; Rios, E.; Canão, P.A.; Velho, S.; Carneiro, F.; Oliveira, M.J.; Cardoso, A.P. Decellularized Colorectal Cancer Matrices as Bioactive Scaffolds for Studying Tumor-Stroma Interactions. Cancers 2022, 14, 359. [Google Scholar] [CrossRef]
- Parmaksiz, M.; Dogan, A.; Odabas, S.; Elçin, A.E.; Elçin, Y.M. Clinical Applications of Decellularized Extracellular Matrices for Tissue Engineering and Regenerative Medicine. Biomed. Mater. 2016, 11, 22003. [Google Scholar] [CrossRef]
- Kim, B.S.; Kim, H.; Gao, G.; Jang, J.; Cho, D.-W. Decellularized Extracellular Matrix: A Step towards the next Generation Source for Bioink Manufacturing. Biofabrication 2017, 9, 34104. [Google Scholar] [CrossRef]
- Schüssler, M.H.; Skoudy, A.; Ramaekers, F.; Real, F.X. Intermediate Filaments as Differentiation Markers of Normal Pancreas and Pancreas Cancer. Am. J. Pathol. 1992, 140, 559–568. [Google Scholar]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D Collagen Fibrillar Microstructure Guides Pancreatic Cancer Cell Phenotype and Serves as a Critical Design Parameter for Phenotypic Models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [Green Version]
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A Novel Microfluidic 3D Platform for Culturing Pancreatic Ductal Adenocarcinoma Cells: Comparison with in Vitro Cultures and in Vivo Xenografts. Sci. Rep. 2017, 7, 1325. [Google Scholar] [CrossRef] [Green Version]
- Loessner, D.; Rockstroh, A.; Shokoohmand, A.; Holzapfel, B.M.; Wagner, F.; Baldwin, J.; Boxberg, M.; Schmalfeldt, B.; Lengyel, E.; Clements, J.A.; et al. A 3D Tumor Microenvironment Regulates Cell Proliferation, Peritoneal Growth and Expression Patterns. Biomaterials 2019, 190–191, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Hoshiba, T. Decellularized Extracellular Matrix for Cancer Research. Materials 2019, 12, 1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, E.; Natarajan, D.; Sensi, F.; Ajayi, O.; Fassan, M.; Mammano, E.; Pilati, P.; Pavan, P.; Bresolin, S.; Preziosi, M.; et al. Patient-Derived Scaffolds of Colorectal Cancer Metastases as an Organotypic 3D Model of the Liver Metastatic Microenvironment. Cancers 2020, 12, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta, S.; Lonardi, S.; Sensi, F.; D’Angelo, E.; Fassan, M.; Pucciarelli, S.; Valzelli, A.; Biccari, A.; Vermi, W.; Della Bella, C.; et al. Tumor Cells and the Extracellular Matrix Dictate the Pro-Tumoral Profile of Macrophages in CRC. Cancers 2021, 13, 5199. [Google Scholar] [CrossRef]
- Piccoli, M.; D’Angelo, E.; Crotti, S.; Sensi, F.; Urbani, L.; Maghin, E.; Burns, A.; De Coppi, P.; Fassan, M.; Rugge, M.; et al. Decellularized Colorectal Cancer Matrix as Bioactive Microenvironment for in Vitro 3D Cancer Research. J. Cell. Physiol. 2018, 233, 5937–5948. [Google Scholar] [CrossRef]
- Sensi, F.; D’Angelo, E.; Biccari, A.; Marangio, A.; Battisti, G.; Crotti, S.; Fassan, M.; Laterza, C.; Giomo, M.; Elvassore, N.; et al. Establishment of a Human 3D Pancreatic Adenocarcinoma Model Based on a Patient-Derived Extracellular Matrix Scaffold. Transl. Res. 2022, 11, 2945. [Google Scholar] [CrossRef]
- Barbon, S.; Biccari, A.; Stocco, E.; Capovilla, G.; D’Angelo, E.; Todesco, M.; Sandrin, D.; Bagno, A.; Romanato, F.; Macchi, V.; et al. Bio-Engineered Scaffolds Derived from Decellularized Human Esophagus for Functional Organ Reconstruction. Cells 2022, 11, 2945. [Google Scholar] [CrossRef]
- Saldin, L.T.; Klimak, M.; Hill, R.C.; Cramer, M.C.; Huleihel, L.; Li, X.; Quidgley-Martin, M.; Cardenas, D.; Keane, T.J.; Londono, R.; et al. The Effect of Normal, Metaplastic, and Neoplastic Esophageal Extracellular Matrix upon Macrophage Activation. J. Immunol. Regen. Med. 2021, 13, 37. [Google Scholar] [CrossRef]
- Ferreira, L.P.; Gaspar, V.M.; Mendes, L.; Duarte, I.F.; Mano, J.F. Organotypic 3D Decellularized Matrix Tumor Spheroids for High-Throughput Drug Screening. Biomaterials 2021, 275, 120983. [Google Scholar] [CrossRef]
- Leiva, M.C.; Garre, E.; Gustafsson, A.; Svanström, A.; Bogestål, Y.; Håkansson, J.; Ståhlberg, A.; Landberg, G. Breast Cancer Patient-Derived Scaffolds as a Tool to Monitor Chemotherapy Responses in Human Tumor Microenvironments. J. Cell. Physiol. 2021, 236, 4709–4724. [Google Scholar] [CrossRef]
- Franco-Barraza, J.; Raghavan, K.S.; Luong, T.; Cukierman, E. Engineering Clinically-Relevant Human Fibroblastic Cell-Derived Extracellular Matrices. Methods Cell Biol. 2020, 156, 109–160. [Google Scholar] [CrossRef] [PubMed]


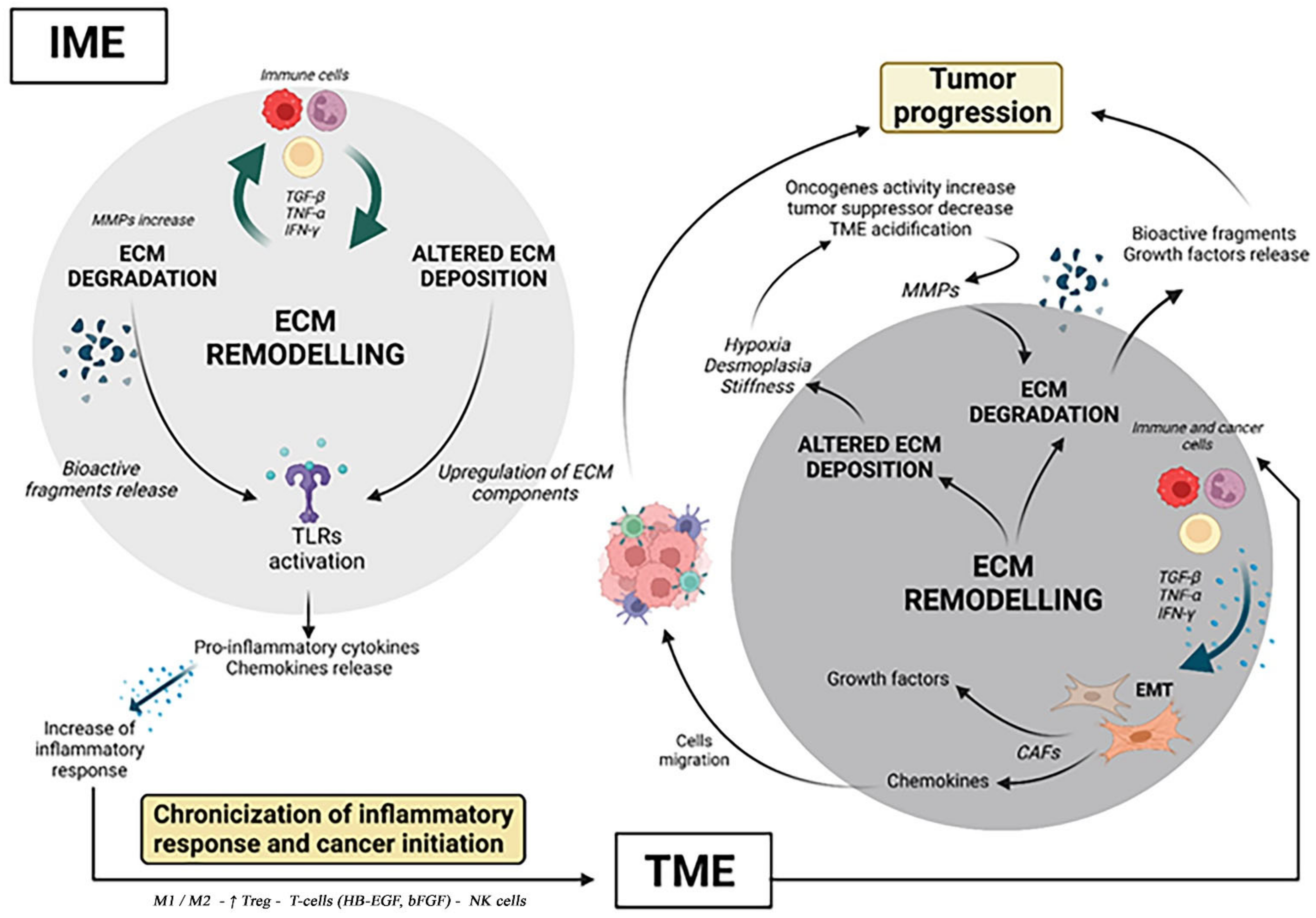{kind=link}
| Components | Functions and Properties |
|---|---|
| Proteins and Structure | |
| Collagens Formed as fibrils within the ECM (Collagen I, II, III, V and XI) | Maintenance of ECM architecture, conferring of mechanical properties [20]. Influence on cell processes such as adhesion and migration. Binding of extracellular growth factors and cytokines [21]. |
| Elastin Composed of single tropoelastin subunits cross-linked | Tissue elasticity, load bearing and storage of mechanical energy [22]. |
| Glycoproteins | |
| Laminins Trimeric structure made of three different chains α, β, γ | Resident in the basement membrane. Cell adhesion, survival, migration, differentiation and migration via integrin [22]. |
| Fibronectin (FN) Arranged into a mesh of fibrils and collagen and is linked to cell surface receptors (integrins) | Binds to collagen, fibrin and glycosaminoglycans, influencing cell adhesion, growth, migration, wound healing and differentiation [23]. |
| Fibrillins | Scaffolds for elastin deposition [23]. |
| GAGs | |
| Hyaluronic acid (HA) | Regulation of numerous cell functions and biological processes through the interaction with cell surface receptors. Lends tissue turgor and facilitates cell migration during tissue morphogenesis and repair [24]. |
| PGs | |
| e.g., Heparan, chondroitin and keratin sulphates. Core of protein domain covalently linked to GAGs. Negatively charged, it can bind water. | Provide compressive resistance to tissue. Reservoir and sequester of growth factors, chemokines and cytokines at the ECM protecting them from degradation and creating effective gradients along ECM [25]. |
| Matricellular proteins | |
| Thrombospondins (TSPs), tenascins (TNs), fibulins, osteopontin (OPN), cartilage oligomeric matrix protein (COMP), CNN family proteins, periostin and R-spondins | Cell behavior modulation, mediation of the interaction between cellular transmembrane receptors and fibrous ECM molecules and key functional regulator of cell-matrix communication [26]. |
| Cancer-associated Endothelial cells |
|
| CAFs |
|
| Cancer-associated adipocytes |
|
| Tumor-associated immune stroma |
|
| dECM Origin | dECM Tissue | Cells Cultured on dECM | Results | Ref. |
|---|---|---|---|---|
| Human | CRC | Human colon adenocarcinoma cells (HT-29—HCT-116) |
| [13] |
| Human | HC—CRC—healthy liver (HL)—CRC and liver metastasis (CRLM) | Human colon adenocarcinoma cells (HT-29 ZsGreen/Luc+ and HCT-116 cells) |
| [233] |
| Human | CRC | Human colon adenocarcinoma cells (HCT-116 and HT-29) |
| [234] |
| Human | Healthy colon and CRC | Human colon adenocarcinoma cells (HT-29 cells) |
| [235] |
| Human | PDAC | Human pancreatic adenocarcinoma cell lines (Panc-1 and AsPC-1) |
| [236] |
| Human | Esophagus from cadavers | Human bone marrow cell line (HM1-SV40) |
| [237] |
| Rat | Esophageal adenocarcinoma (EAC) | Human mononuclear cells (THP-1) and esophageal epithelial cells (Het-1A) |
| [238] |
| Pig | Breast cancer | Human BC cell line (MDA-MB- 231) 3D tumor spheroids Human dermal fibroblasts |
| [239] |
| Human | Breast cancer | Human breast cancer cells (MCF7, T-47D, and MDA-MB-231) |
| [240] |
| Human and murine | Normal and tumor biopsies | Fibroblastic cell derived matrices (fCDM) |
| [241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marangio, A.; Biccari, A.; D’Angelo, E.; Sensi, F.; Spolverato, G.; Pucciarelli, S.; Agostini, M. The Study of the Extracellular Matrix in Chronic Inflammation: A Way to Prevent Cancer Initiation? Cancers 2022, 14, 5903. https://doi.org/10.3390/cancers14235903
Marangio A, Biccari A, D’Angelo E, Sensi F, Spolverato G, Pucciarelli S, Agostini M. The Study of the Extracellular Matrix in Chronic Inflammation: A Way to Prevent Cancer Initiation? Cancers. 2022; 14(23):5903. https://doi.org/10.3390/cancers14235903
Chicago/Turabian StyleMarangio, Asia, Andrea Biccari, Edoardo D’Angelo, Francesca Sensi, Gaya Spolverato, Salvatore Pucciarelli, and Marco Agostini. 2022. "The Study of the Extracellular Matrix in Chronic Inflammation: A Way to Prevent Cancer Initiation?" Cancers 14, no. 23: 5903. https://doi.org/10.3390/cancers14235903