
Prognostic Significance of Cuproptosis-Related Gene Signatures in Breast Cancer Based on Transcriptomic Data Analysis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Download and Preprocessing
2.2. Acquisition of Differentially Expressed Genes Related to Cuproptosis
2.3. Identification of Breast Cancer Subtypes Based on Different Cuproptosis-Related Genes
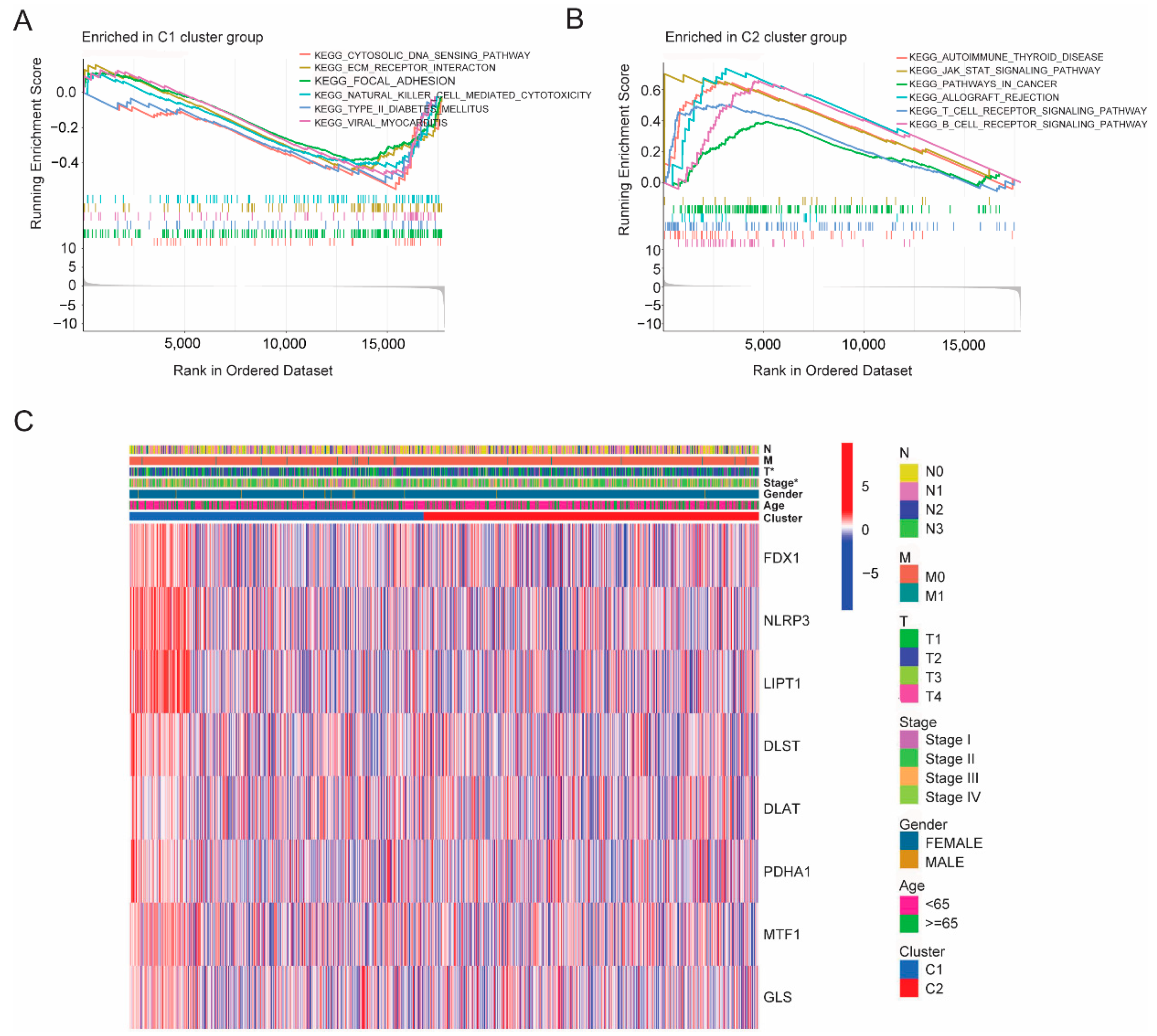
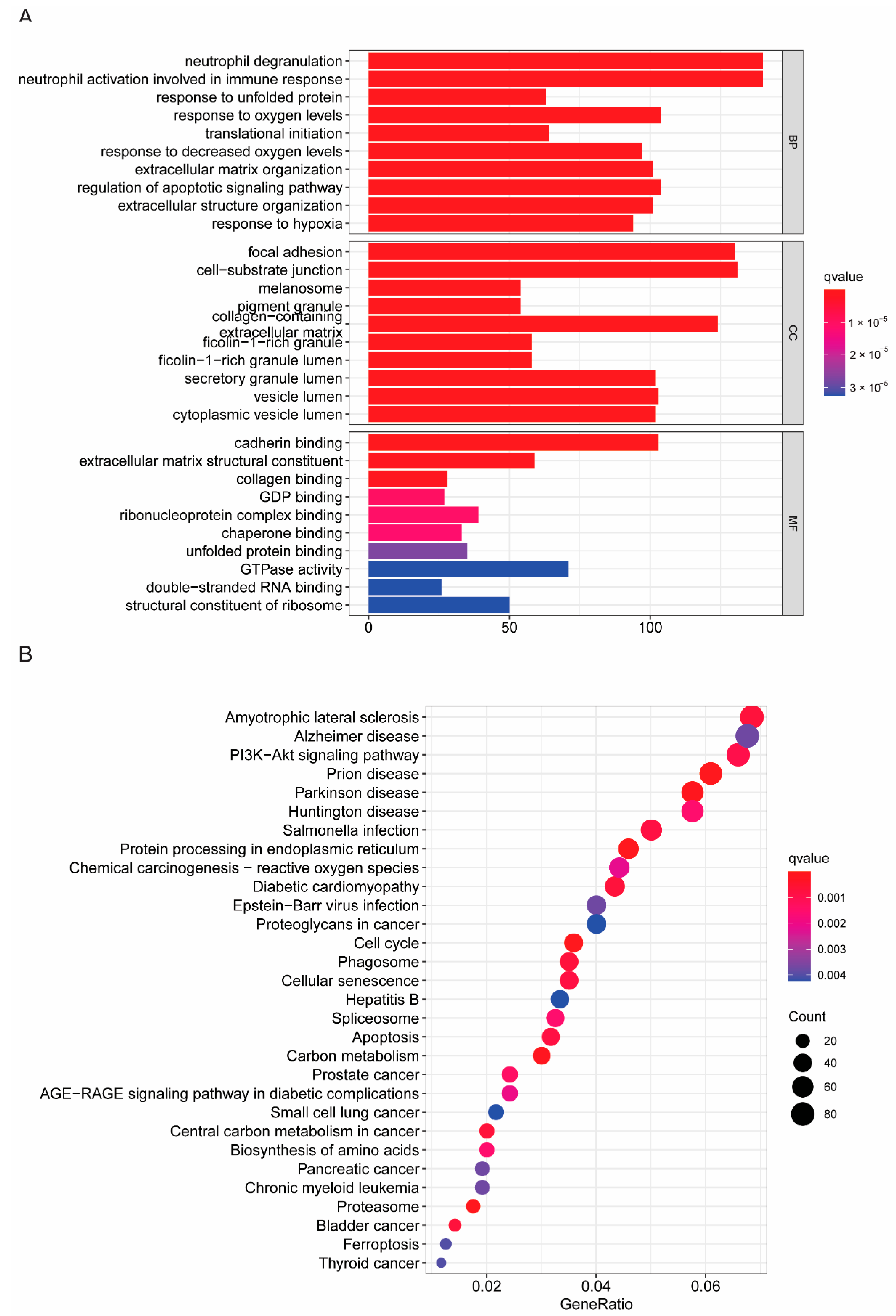
2.4. Identification Differential Expression Gene and GSEA Functional Analysis between the Subtypes
2.5. Construction and Validation of the CRGs Prognostic Model
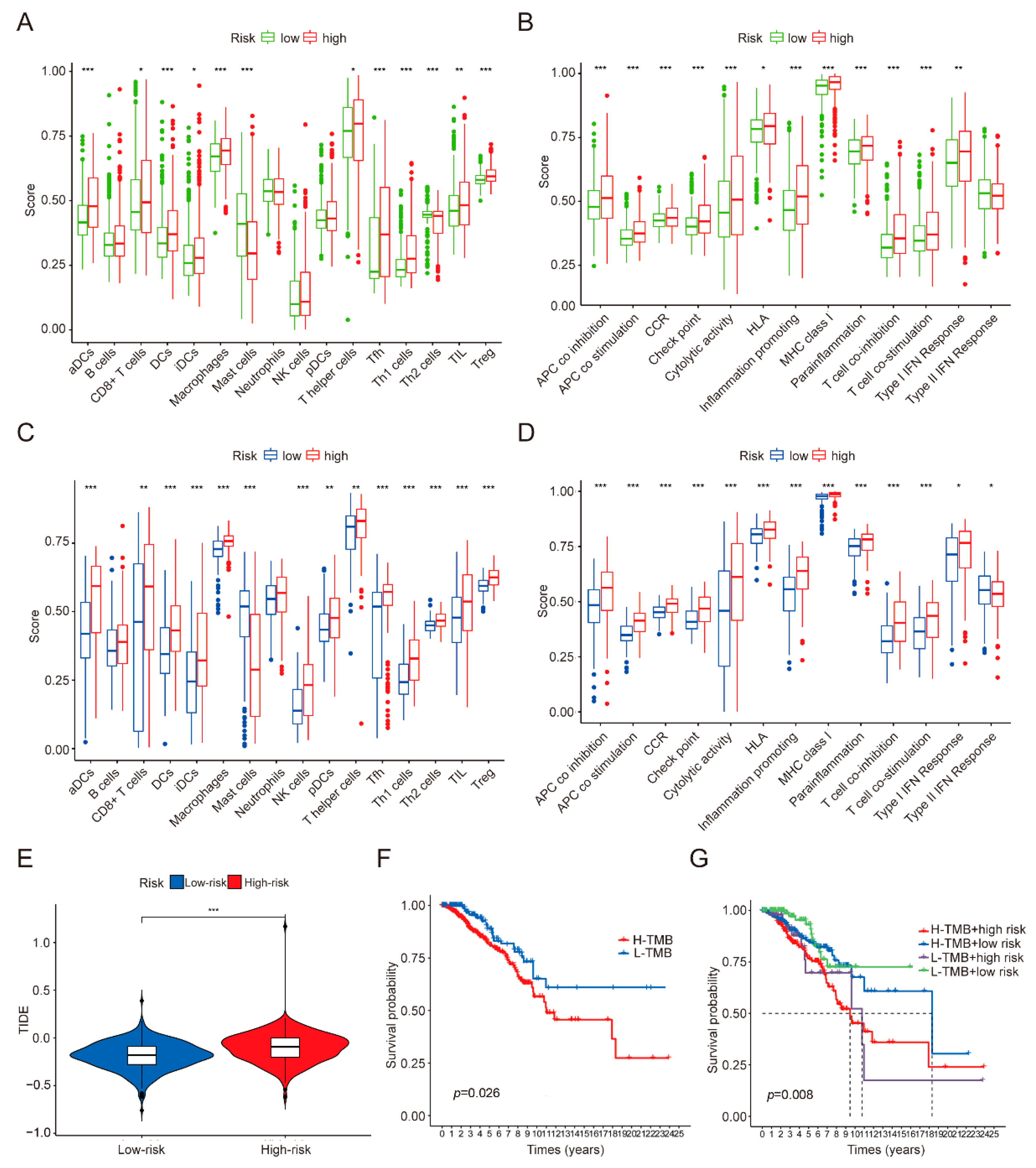
2.6. Estimation of Immune Cell Infiltration Based on the CRGs Model
2.7. Construction of Disease Prediction Models and Screening of Independent Prognostic Factors among the CRGs Risk Score and Clinical Indicators
2.8. Statistical Analyses
3. Results
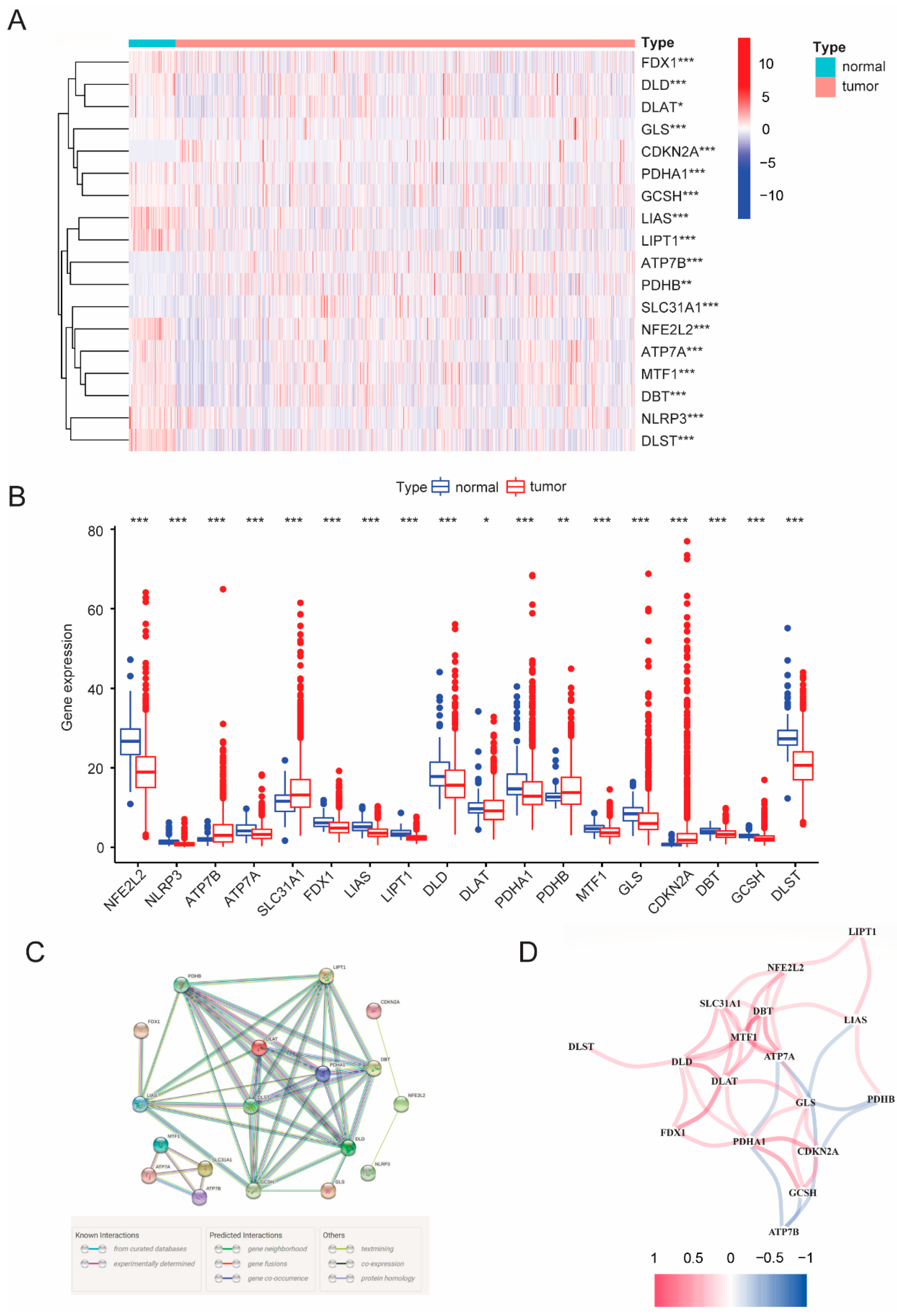
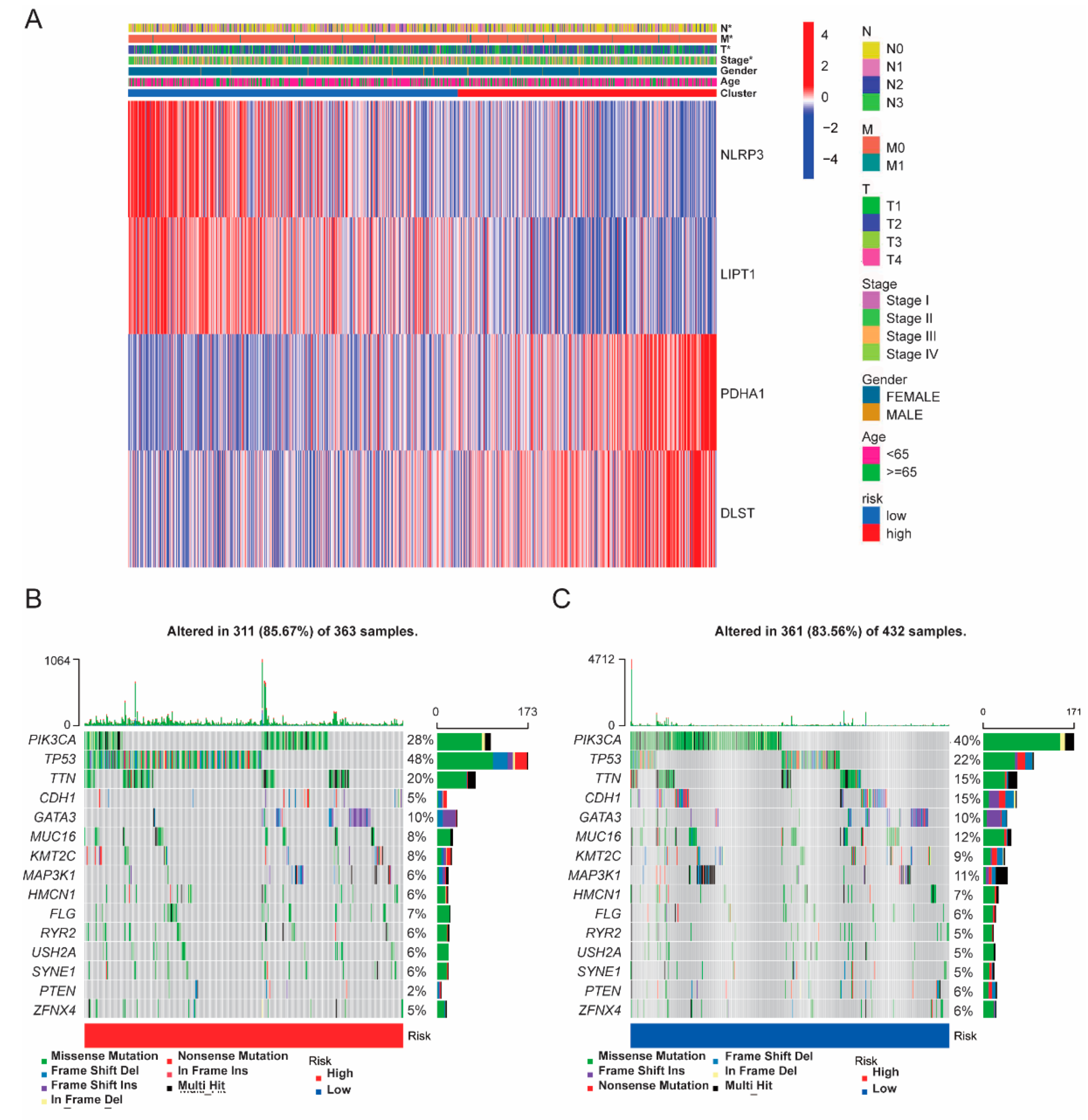
3.1. Differential CRGs Expression between Para-Carcinoma Tissues and Tumor Tissues
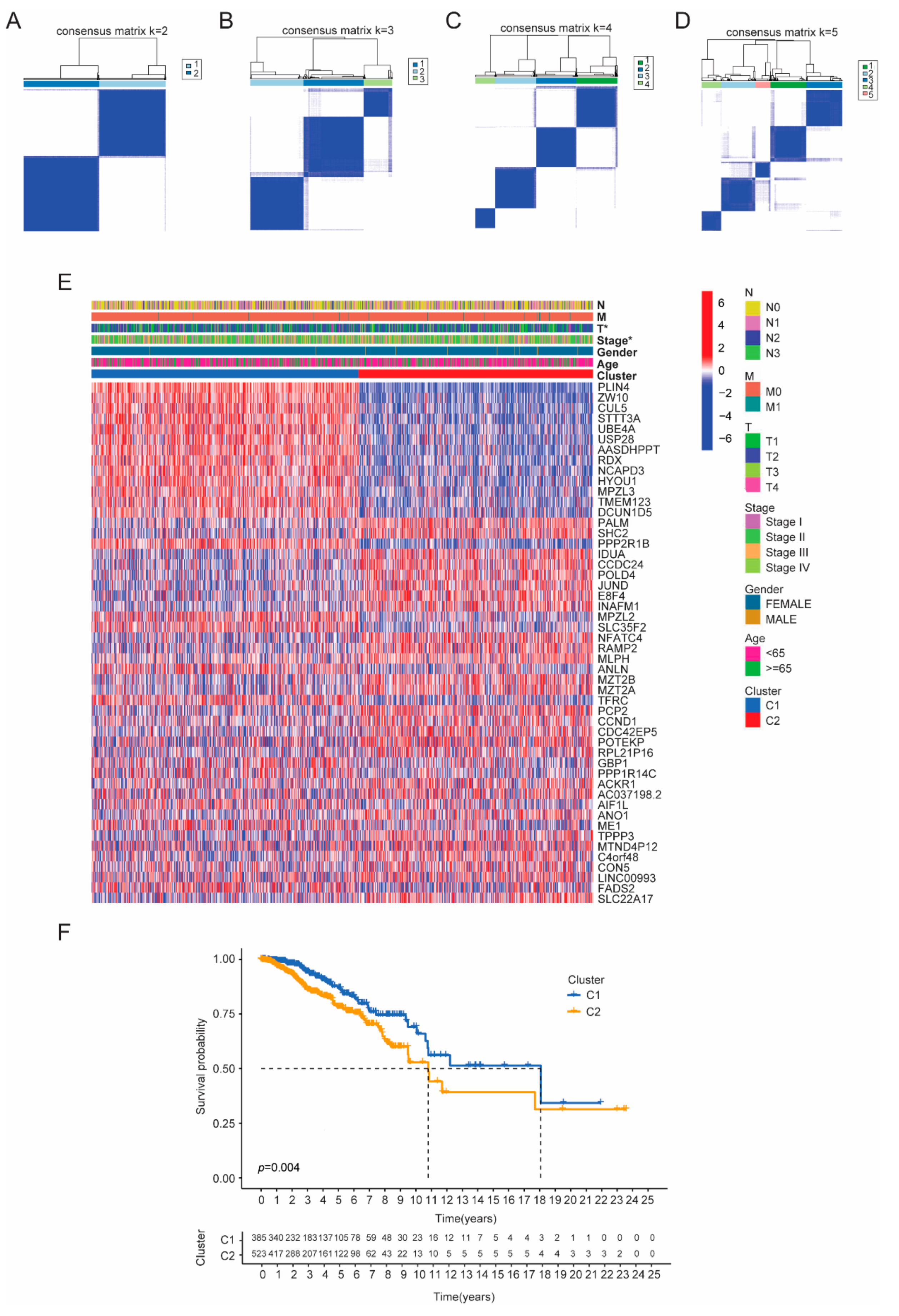
3.2. Identifying Tumor Subtypes Based on Differentially Expressed CRGs
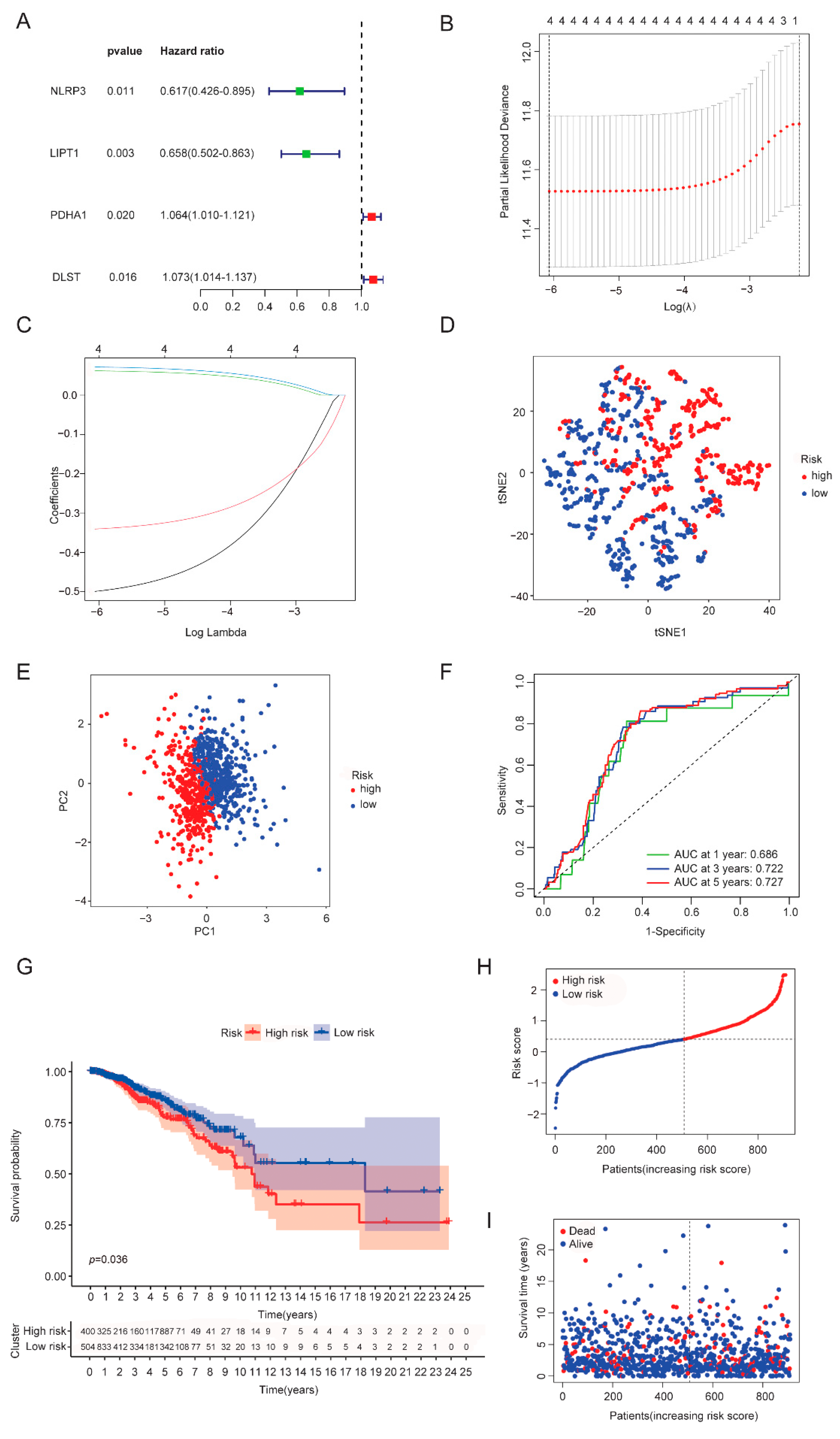
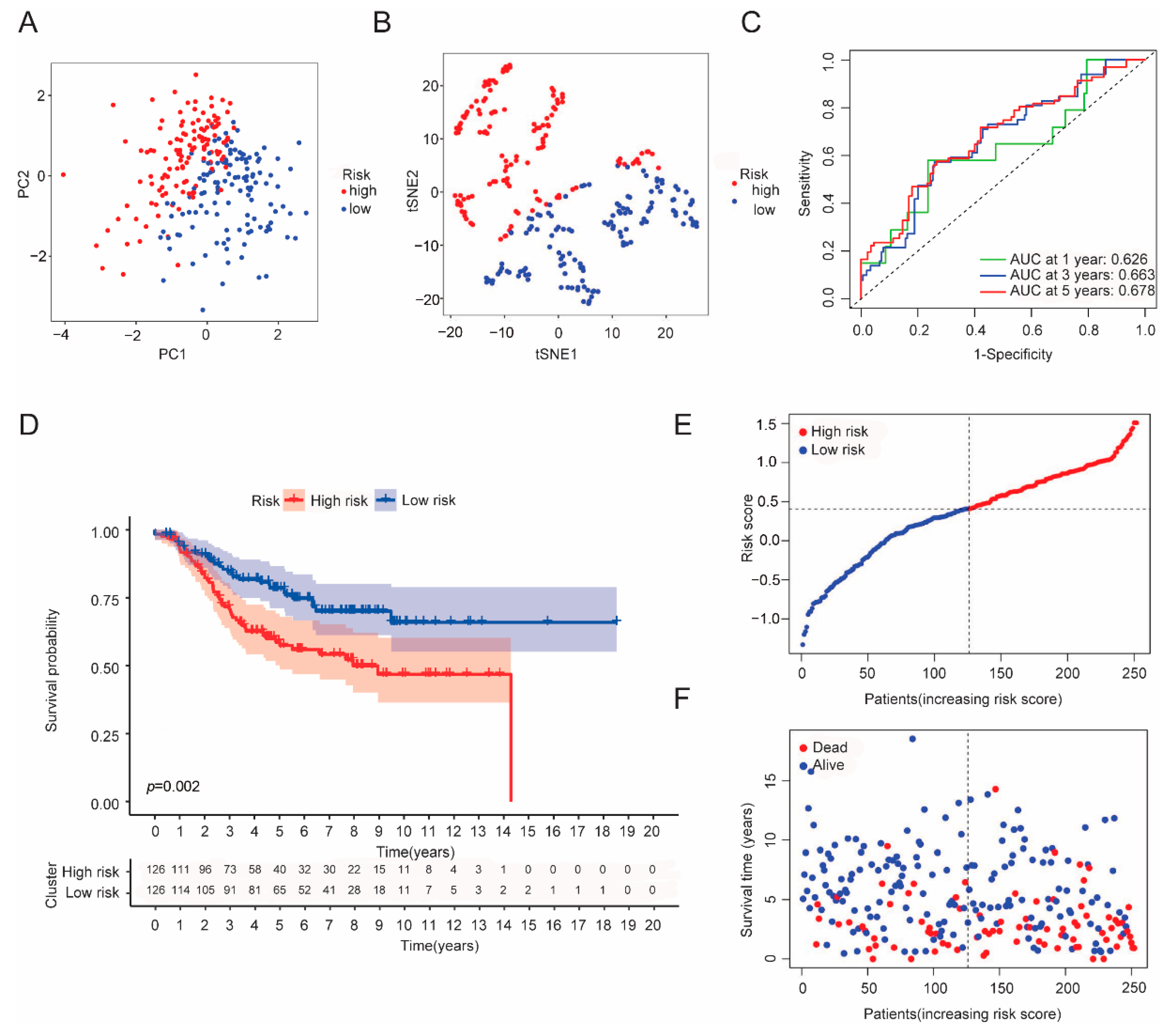
3.3. Establishment of a CRGs Risk Model
3.4. Somatic Mutation and Functional Analysis Based on Risk Subgroups
3.5. Immune Infiltration, Immune Evasion and Tumor Mutational Burden Analysis Based on the Riskscore Subgroups
3.6. Independent Factor Prognostic Value of the Risk Model and Construction of the Nomogram
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nikolov, I.; Kostev, K.; Kalder, M. Incidence of other cancer diagnoses in women with breast cancer: A retrospective cohort study with 42,248 women. Breast Cancer Res. Treat. 2022, 195, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.L.; Huang, T.; Wu, B.L.; He, W.X.; Liu, D. Stem cells in cancer therapy: Opportunities and challenges. Oncotarget 2017, 8, 75756–75766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, C.; Su, X. Emerging impact of the long noncoding RNA MIR22HG on proliferation and apoptosis in multiple human cancers. J. Exp. Clin. Cancer Res. 2020, 39, 271. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, X.; Liu, N.; Shi, Y.; Liu, Y.; Ouyang, L.; Tam, S.; Xiao, D.; Liu, S.; Wen, F.; et al. A Nuclear Long Non-Coding RNA LINC00618 Accelerates Ferroptosis in a Manner Dependent upon Apoptosis. Mol. Ther. 2021, 29, 263–274. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zheng, J.; Hu, J.; Chen, J.; Wei, X. Long non-coding RNAs and pyroptosis. Clin. Chim. Acta 2020, 504, 201–208. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Liu, T.; Wang, J. Radiosensitization-Related Cuproptosis LncRNA Signature in Non-Small Cell Lung Cancer. Genes 2022, 13, 2080. [Google Scholar] [CrossRef]
- Zhang, N.; Zhao, Y.D.; Wang, X.M. CXCL10 an important chemokine associated with cytokine storm in COVID-19 infected patients. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7497–7505. [Google Scholar] [CrossRef]
- Yin, F.; Wang, S.; Hou, C.; Zhang, Y.; Yang, Z.; Wang, X. Development and validation of nomograms for predicting overall survival and cancer specific survival in locally advanced breast cancer patients: A SEER population-based study. Front. Public Health 2022, 10, 969030. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Fan, R.; Xie, L. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes 2022, 13, 851. [Google Scholar] [CrossRef] [PubMed]
- Kahlson, M.A.; Dixon, S.J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Chen, Y.; Pang, D. Identification of cuproptosis-related patterns and construction of a scoring system for predicting prognosis, tumor microenvironment-infiltration characteristics, and immunotherapy efficacy in breast cancer. Front. Oncol. 2022, 12, 966511. [Google Scholar] [CrossRef]
- Li, J.; Wu, F.; Li, C.; Sun, S.; Feng, C.; Wu, H.; Chen, X.; Wang, W.; Zhang, Y.; Liu, M.; et al. The cuproptosis-related signature predicts prognosis and indicates immune microenvironment in breast cancer. Front. Genet. 2022, 13, 977322. [Google Scholar] [CrossRef]
- Huang, P.; Li, F.; Li, L.; You, Y.; Luo, S.; Dong, Z.; Gao, Q.; Wu, S.; Brünner, N.; Stenvang, J. lncRNA profile study reveals the mRNAs and lncRNAs associated with docetaxel resistance in breast cancer cells. Sci. Rep. 2018, 8, 17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uriu-Adams, J.Y.; Keen, C.L. Copper, oxidative stress, and human health. Mol. Asp. Med. 2005, 26, 268–298. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Ramchandani, D.; Vahdat, L. Copper Depletion as a Therapeutic Strategy in Cancer. Met. Ions Life Sci. 2019, 19. [Google Scholar] [CrossRef]
- Gou, Y.; Chen, M.; Li, S.; Deng, J.; Li, J.; Fang, G.; Yang, F.; Huang, G. Dithiocarbazate-Copper Complexes for Bioimaging and Treatment of Pancreatic Cancer. J. Med. Chem. 2021, 64, 5485–5499. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 6836–6841. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Wu, Y.; Liu, Z.; Yu, Y.; Sun, S.; Guo, M.; Sun, B.; Huang, C. CDKN2A-mediated molecular subtypes characterize the hallmarks of tumor microenvironment and guide precision medicine in triple-negative breast cancer. Front. Immunol. 2022, 13, 970950. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Meylan, E.; Tschopp, J. Intracellular pattern-recognition receptors. Adv. Drug Deliv. Rev. 2008, 60, 830–840. [Google Scholar] [CrossRef]
- Plato, A.; Hardison, S.E.; Brown, G.D. Pattern recognition receptors in antifungal immunity. Semin. Immunopathol. 2015, 37, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef]
- Liu, C.; He, D.; Zhang, S.; Chen, H.; Zhao, J.; Li, X.; Zeng, X. Homogeneous Polyporus Polysaccharide Inhibit Bladder Cancer by Resetting Tumor-Associated Macrophages Toward M1 Through NF-κB/NLRP3 Signaling. Front. Immunol. 2022, 13, 839460. [Google Scholar] [CrossRef]
- Faria, S.S.; Costantini, S.; de Lima, V.C.C.; de Andrade, V.P.; Rialland, M.; Cedric, R.; Budillon, A.; Magalhães, K.G. NLRP3 inflammasome-mediated cytokine production and pyroptosis cell death in breast cancer. J. Biomed. Sci. 2021, 28, 26. [Google Scholar] [CrossRef]
- Tran, V.T.; Pham, D.V.; Choi, D.Y.; Park, P.H. Mitophagy Induction and Aryl Hydrocarbon Receptor-Mediated Redox Signaling Contribute to the Suppression of Breast Cancer Cell Growth by Taloxifene via Regulation of Inflammasomes Activation. Antioxid. Redox Signal. 2022, 37, 1030–1050. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Liu, X.; Zeng, X.; Liu, Y.; Zhang, C.; Zhang, Q.; Xu, J. Comprehensive Analysis of Cuproptosis-Related Genes in Immune Infiltration and Prognosis in Melanoma. Front. Pharmacol. 2022, 13, 930041. [Google Scholar] [CrossRef]
- Deng, L.; Jiang, A.; Zeng, H.; Peng, X.; Song, L. Comprehensive analyses of PDHA1 that serves as a predictive biomarker for immunotherapy response in cancer. Front. Pharmacol. 2022, 13, 947372. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yu, M.; Fei, B.; Fang, X.; Ma, T.; Wang, D. miR-21-5p targets PDHA1 to regulate glycolysis and cancer progression in gastric cancer. Oncol. Rep. 2018, 40, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, X.; Ji, Y.; Li, X.; Li, Y.; Yu, D.; Yuan, Y.; Liu, J.; Li, H.; Zhang, M.; et al. Pyruvate dehydrogenase expression is negatively associated with cell stemness and worse clinical outcome in prostate cancers. Oncotarget 2017, 8, 13344–13356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, N.; Spurlin, G.; Korm, S.; Huang, S.; Anderson, N.M.; Huiting, L.N.; Liu, H.; Feng, H. α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting. Cancers 2022, 14, 2983. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Qin, X.; Finan, J.M.; Lam, A.; Athoe, J.; Missiaen, R.; Skuli, N.; Kennedy, A.; Saini, A.S.; Tao, T.; et al. Metabolic Enzyme DLST Promotes Tumor Aggression and Reveals a Vulnerability to OXPHOS Inhibition in High-Risk Neuroblastoma. Cancer Res. 2021, 81, 4417–4430. [Google Scholar] [CrossRef]
- Gu, Y.; Zhang, P.; Wang, J.; Lin, C.; Liu, H.; Li, H.; He, H.; Li, R.; Zhang, H.; Zhang, W. Somatic ARID1A mutation stratifies patients with gastric cancer to PD-1 blockade and adjuvant chemotherapy. Cancer Immunol. Immunother. 2022. [Google Scholar] [CrossRef]
- Wang, H.L.; Liu, P.F.; Yue, J.; Jiang, W.H.; Cui, Y.L.; Ren, H.; Wang, H.; Zhuang, Y.; Liu, Y.; Jiang, D.; et al. Somatic gene mutation signatures predict cancer type and prognosis in multiple cancers with pan-cancer 1000 gene panel. Cancer Lett. 2020, 470, 181–190. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Pagès, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Annunziata, F.; Sirvinskas, D.; Omrani, O.; Li, H.; Rasa, S.M.M.; Krepelova, A.; Adam, L.; Neri, F. Establishment and evaluation of module-based immune-associated gene signature to predict overall survival in patients of colon adenocarcinoma. J. Biomed. Sci. 2022, 29, 81. [Google Scholar] [CrossRef] [PubMed]
- Ran, G.H.; Lin, Y.Q.; Tian, L.; Zhang, T.; Yan, D.M.; Yu, J.H.; Deng, Y.C. Natural killer cell homing and trafficking in tissues and tumors: From biology to application. Signal Transduct. Target. Ther. 2022, 7, 205. [Google Scholar] [CrossRef]
- Lelièvre, P.; Sancey, L.; Coll, J.L.; Deniaud, A.; Busser, B. The Multifaceted Roles of Copper in Cancer: A Trace Metal Element with Dysregulated Metabolism, but Also a Target or a Bullet for Therapy. Cancers 2020, 12, 3594. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Deng, J.; Pan, T.; Zhu, Z.; Zhou, X.; Lv, C.; Li, H.; Peng, W.; Lin, B.; Cai, C.; et al. Prognostic Significance of Cuproptosis-Related Gene Signatures in Breast Cancer Based on Transcriptomic Data Analysis. Cancers 2022, 14, 5771. https://doi.org/10.3390/cancers14235771
Zhou Z, Deng J, Pan T, Zhu Z, Zhou X, Lv C, Li H, Peng W, Lin B, Cai C, et al. Prognostic Significance of Cuproptosis-Related Gene Signatures in Breast Cancer Based on Transcriptomic Data Analysis. Cancers. 2022; 14(23):5771. https://doi.org/10.3390/cancers14235771
Chicago/Turabian StyleZhou, Zizhen, Jinhai Deng, Teng Pan, Zhengjie Zhu, Xiulan Zhou, Chunxin Lv, Huanxin Li, Weixiong Peng, Bihai Lin, Cuidan Cai, and et al. 2022. "Prognostic Significance of Cuproptosis-Related Gene Signatures in Breast Cancer Based on Transcriptomic Data Analysis" Cancers 14, no. 23: 5771. https://doi.org/10.3390/cancers14235771