
Identification of Immunogenic Cell Death-Related Signature for Glioma to Predict Survival and Response to Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
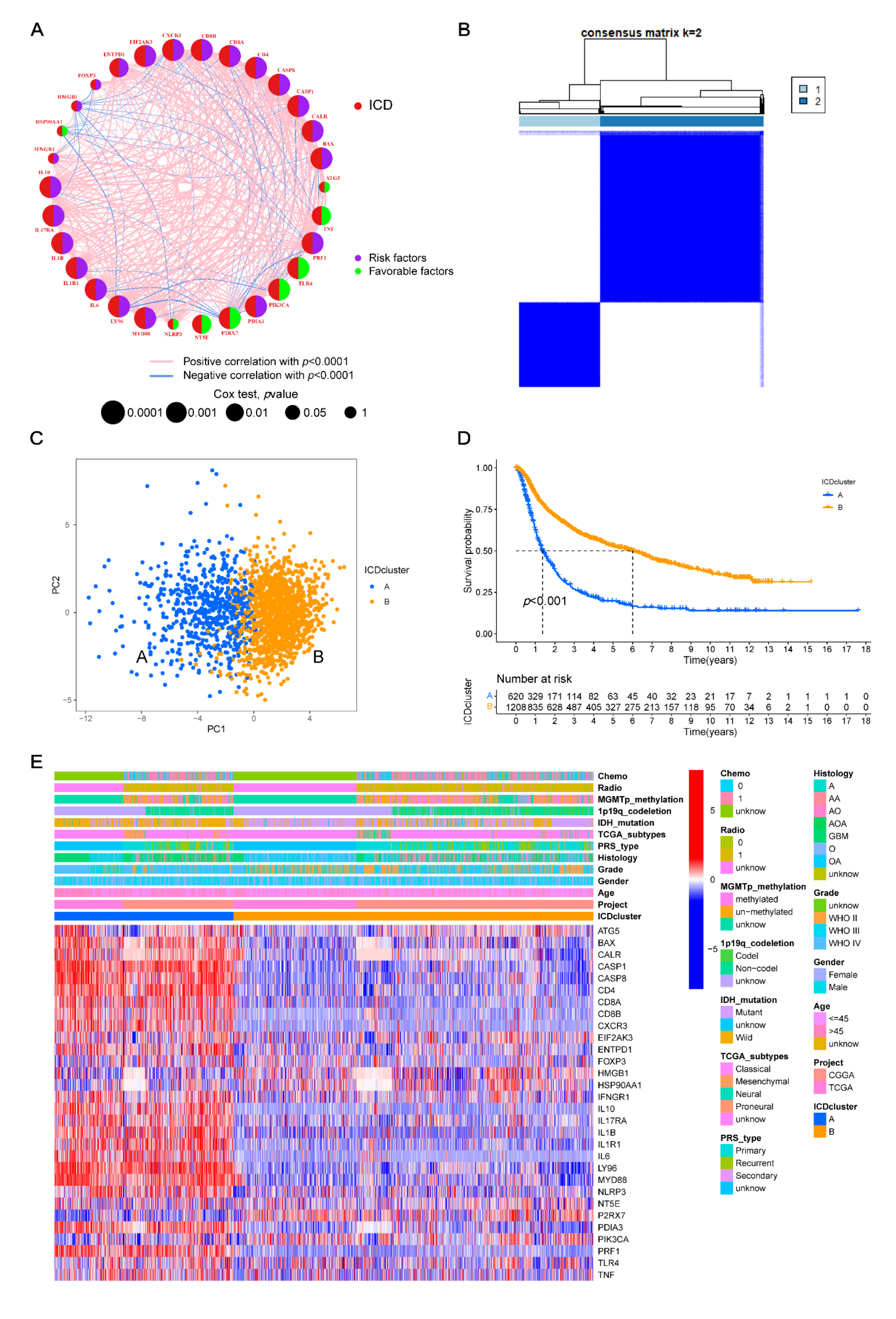
2.2. Consensus Clustering
2.3. Glioma in Different Molecular Clusters: Clinical Features and Prognosis
2.4. Correlations of Molecular Clusters with TME and Immune Checkpoint Genes
2.5. Differentially Expressed Genes (DEGs) Identification and Functional Enrichment Analysis
2.6. Development of an ICD-Related Risk-Score Model
2.7. Tissue Samples
2.8. RNA Extracting and Quantitative Real-Time PCR
2.9. Clinical Relevance and Stratified Analysis of the Risk-Score
2.10. Immune Status and Cancer Stem Cell (CSC) Index in Different Risk Groups
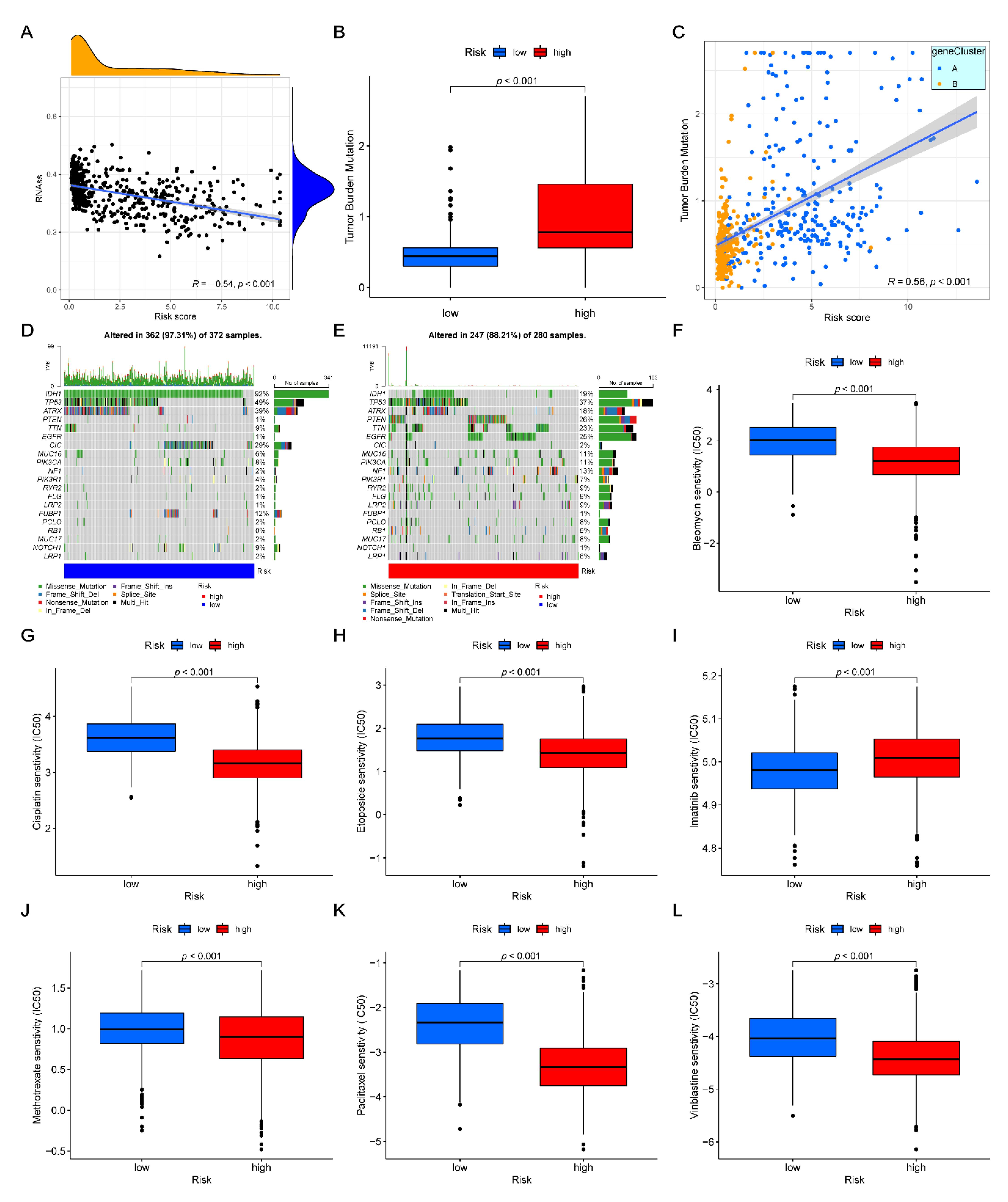
2.11. Drug Sensitivity and Mutation Analysis
2.12. Development of the Nomogram
2.13. Statistical Analyses
3. Results
3.1. Transcriptional Changes of ICD-Related Genes in Glioma
3.2. Construction of ICD-Related Gene Clusters in Glioma
3.3. TME Characteristics Differ between Clusters
3.4. Identifying Gene Clusters
3.5. Construction of the Prognostic Risk Score
3.6. Verification of the Expression Levels of 14-Signature Genes
3.7. Clinical Relevance and Prognostic Risk-Score Stratification Analyses
3.8. Analyzing the Immune Checkpoint and TME between the Risk Subgroups
3.9. Correlation between CSC Index and Risk-Score
3.10. Drug Sensitivity and Mutation Analysis
3.11. Constructing a Prognostic Nomogram
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garg, A.D.; Agostinis, P. ER Stress, Autophagy and Immunogenic Cell Death in Photodynamic Therapy-Induced Anti-Cancer Immune Responses. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2014, 13, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus Guidelines for the Detection of Immunogenic Cell Death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1beta-Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic Death of Colon Cancer Cells Treated with Oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding Cell Death Signals in Inflammation and Immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Séror, C.; Métivier, D.; Perfettini, J.-L.; Zitvogel, L.; et al. Chemotherapy Induces ATP Release from Tumor Cells. Cell Cycle Georget. Tex 2009, 8, 3723–3728. [Google Scholar] [CrossRef]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac Glycosides Exert Anticancer Effects by Inducing Immunogenic Cell Death. Sci. Transl. Med. 2012, 4, 143ra99. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER Stress and Immunogenic Cell Death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2010-2014. Neuro–Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary Brain Tumours in Adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination with Immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Feng, S.; Liang, X.; Li, J.; Wang, Z.; Zhang, H.; Dai, Z.; Luo, P.; Liu, Z.; Zhang, J.; Xiao, X.; et al. Immunogenic Cell Death Related Risk Model to Delineate Ferroptosis Pathway and Predict Immunotherapy Response of Patients with GBM. Front. Immunol. 2022, 13, 992855. [Google Scholar] [CrossRef]
- Cai, J.; Hu, Y.; Ye, Z.; Ye, L.; Gao, L.; Wang, Y.; Sun, Q.; Tong, S.; Yang, J.; Chen, Q. Immunogenic Cell Death-Related Risk Signature Predicts Prognosis and Characterizes the Tumour Microenvironment in Lower-Grade Glioma. Front. Immunol. 2022, 13, 1011757. [Google Scholar] [CrossRef]
- Garg, A.D.; De Ruysscher, D.; Agostinis, P. Immunological Metagene Signatures Derived from Immunogenic Cancer Cell Death Associate with Improved Survival of Patients with Lung, Breast or Ovarian Malignancies: A Large-Scale Meta-Analysis. Oncoimmunology 2015, 5, e1069938. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Gonen, M.; Smith, J.J.; DeMatteo, R.P. Nomograms in Oncology: More than Meets the Eye. Lancet Oncol. 2015, 16, e173–e180. [Google Scholar] [CrossRef]
- Laird, P.W. The Power and the Promise of DNA Methylation Markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A Census of Human Transcription Factors: Function, Expression and Evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef]
- Snyder, A.; Wolchok, J.D.; Chan, T.A. Genetic Basis for Clinical Response to CTLA-4 Blockade. N. Engl. J. Med. 2015, 372, 783. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Gupta, G.; Borglum, K.; Chen, H. Immunogenic Cell Death: A Step Ahead of Autophagy in Cancer Therapy. J. Cancer Immunol. 2021, 3, 47–59. [Google Scholar] [CrossRef]
- Li, D.; Patel, C.B.; Xu, G.; Iagaru, A.; Zhu, Z.; Zhang, L.; Cheng, Z. Visualization of Diagnostic and Therapeutic Targets in Glioma with Molecular Imaging. Front. Immunol. 2020, 11, 592389. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Lo, K.-W.; Lui, V.W.Y. Omics-Wide Quantitative B-Cell Infiltration Analyses Identify GPR18 for Human Cancer Prognosis with Superiority over CD20. Commun. Biol. 2020, 3, 234. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil Infiltration into Human Gliomas. Acta Neuropathologica 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangère, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood Baseline Neutrophil Count Predicts Bevacizumab Efficacy in Glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-Infiltrated Innate Immune Cells Resemble M0 Macrophage Phenotype. JCI Insight 2016, 1, 85841. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., II; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased Regulatory T-Cell Fraction Amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients with Malignant Glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef]
- Fecci, P.E.; Sweeney, A.E.; Grossi, P.M.; Nair, S.K.; Learn, C.A.; Mitchell, D.A.; Cui, X.; Cummings, T.J.; Bigner, D.D.; Gilboa, E.; et al. Systemic Anti-CD25 Monoclonal Antibody Administration Safely Enhances Immunity in Murine Glioma without Eliminating Regulatory T Cells. Clin. Cancer Res. 2006, 12, 4294–4305. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The Role of Human Glioma-Infiltrating Microglia/Macrophages in Mediating Antitumor Immune Responses1. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B Cells and Tertiary Lymphoid Structures as Determinants of Tumour Immune Contexture and Clinical Outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-Programmed Nutrient Partitioning in the Tumour Microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and Potential of PD-1/PD-L1 Checkpoint Blockade Immunotherapy for Glioblastoma. J. Exp. Clin. Cancer Res. CR 2019, 38, 87. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 Expression and Prognostic Impact in Glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef]
- Rotte, A.; D’Orazi, G.; Bhandaru, M. Nobel Committee Honors Tumor Immunologists. J. Exp. Clin. Cancer Res. CR 2018, 37, 262. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Id | HR | 95%CI | p-Value |
|---|---|---|---|
| BAX | 1.1447 | 1.0528~1.2447 | 0.0016 |
| CALR | 1.1871 | 1.0917~1.2909 | p < 0.001 |
| CASP1 | 1.1335 | 1.0664~1.2049 | p < 0.001 |
| CASP8 | 1.2092 | 1.1051~1.3232 | p < 0.001 |
| CD4 | 1.1065 | 1.0368~1.1808 | 0.0023 |
| IL10 | 1.1363 | 1.0367~1.2455 | 0.0063 |
| IL1R1 | 1.0916 | 1.0307~1.1560 | 0.0028 |
| IL6 | 1.0610 | 1.0134~1.1109 | 0.0115 |
| LY96 | 1.0940 | 1.0451~1.1453 | p < 0.001 |
| MYD88 | 1.2355 | 1.1433~1.3351 | p < 0.001 |
| NT5E | 0.8990 | 0.8479~0.9531 | p < 0.001 |
| PDIA3 | 1.2042 | 1.0773~1.3459 | 0.0011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Jiang, H.; Yan, T.; Deng, G.; Chen, Q. Identification of Immunogenic Cell Death-Related Signature for Glioma to Predict Survival and Response to Immunotherapy. Cancers 2022, 14, 5665. https://doi.org/10.3390/cancers14225665
Sun Z, Jiang H, Yan T, Deng G, Chen Q. Identification of Immunogenic Cell Death-Related Signature for Glioma to Predict Survival and Response to Immunotherapy. Cancers. 2022; 14(22):5665. https://doi.org/10.3390/cancers14225665
Chicago/Turabian StyleSun, Zhiqiang, Hongxiang Jiang, Tengfeng Yan, Gang Deng, and Qianxue Chen. 2022. "Identification of Immunogenic Cell Death-Related Signature for Glioma to Predict Survival and Response to Immunotherapy" Cancers 14, no. 22: 5665. https://doi.org/10.3390/cancers14225665