
ERK Inhibitor Ulixertinib Inhibits High-Risk Neuroblastoma Growth In Vitro and In Vivo
 , , , , , , , , and
, , , , , , , , and 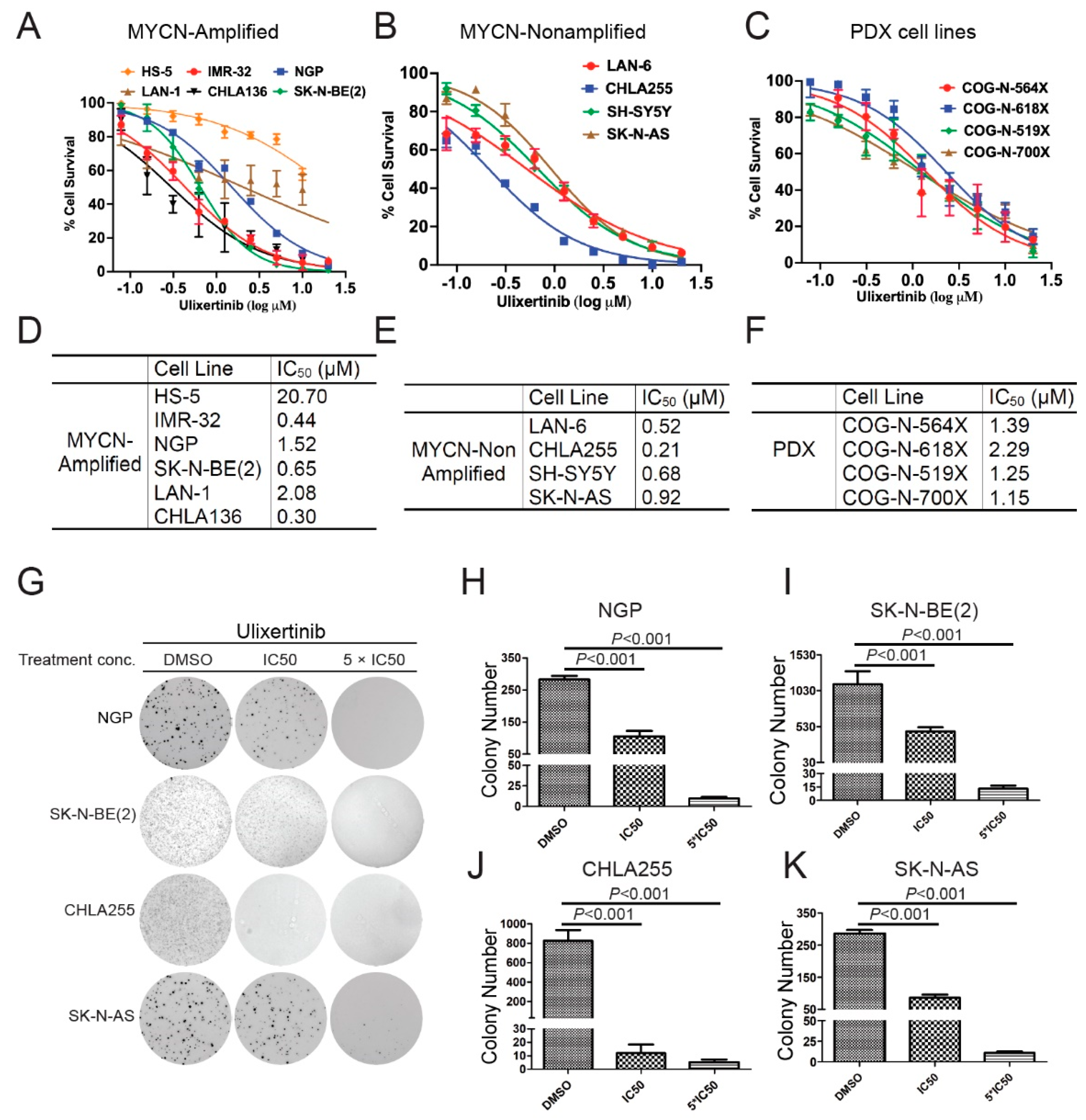{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. NB Cell Lines and Patient-Derived Xenograft Cells
2.3. Cell Proliferation Assay
2.4. Anchorage-Independent Colony Formation Assay
2.5. Immunoblotting
2.6. Synergy Studies
2.7. NB Xenograft Mouse Model
2.8. RNA-Seq Analysis
2.9. Mass Spectrometry Data Analysis
2.10. Statistical and Bioinformatics Analysis
3. Results
3.1. Ulixertinib Significantly Inhibits NB Cell Proliferation
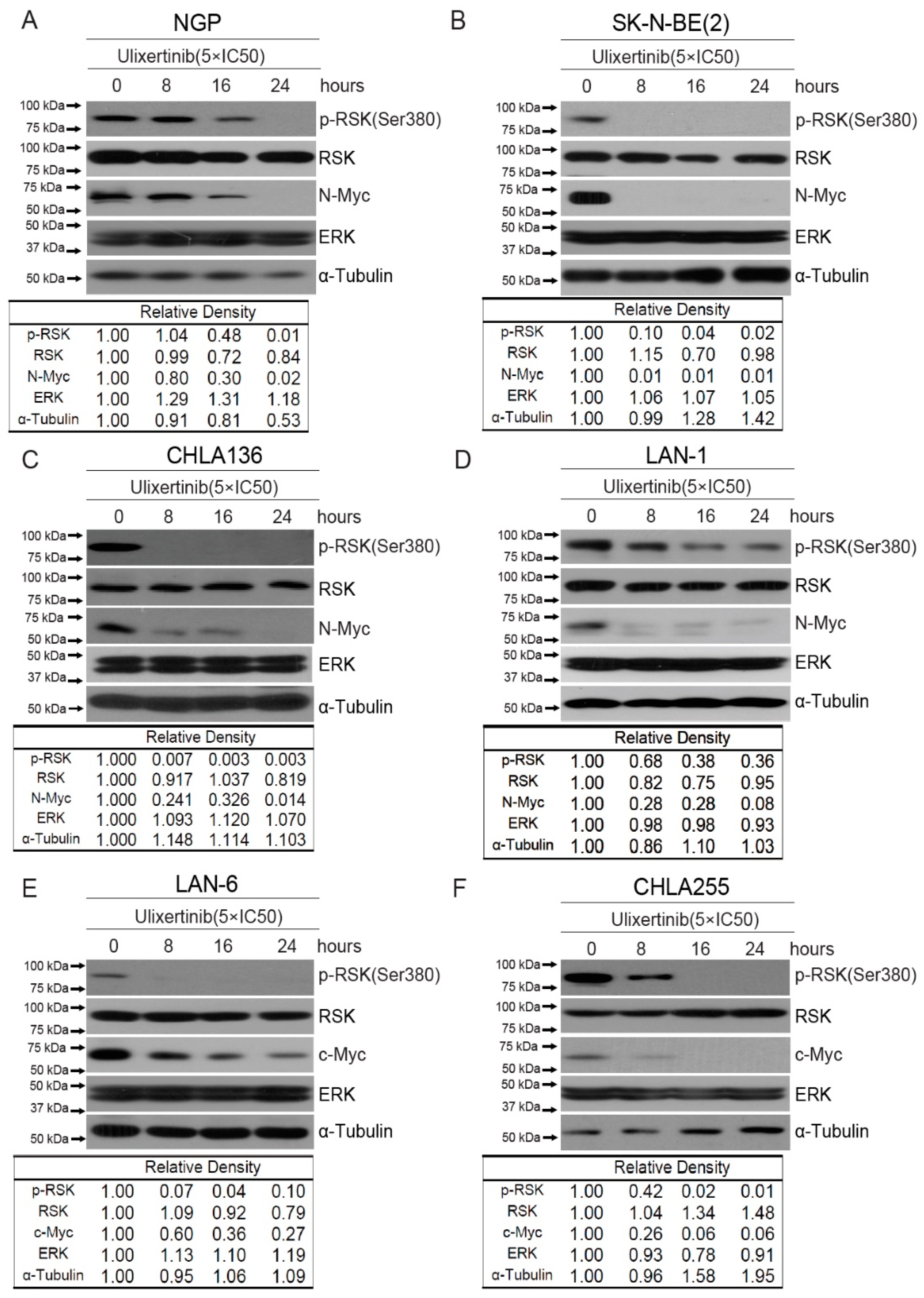
3.2. Ulixertinib Inhibits RSK Phosphorylation and Downregulates c-Myc/N-Myc Protein Levels in NB Cells
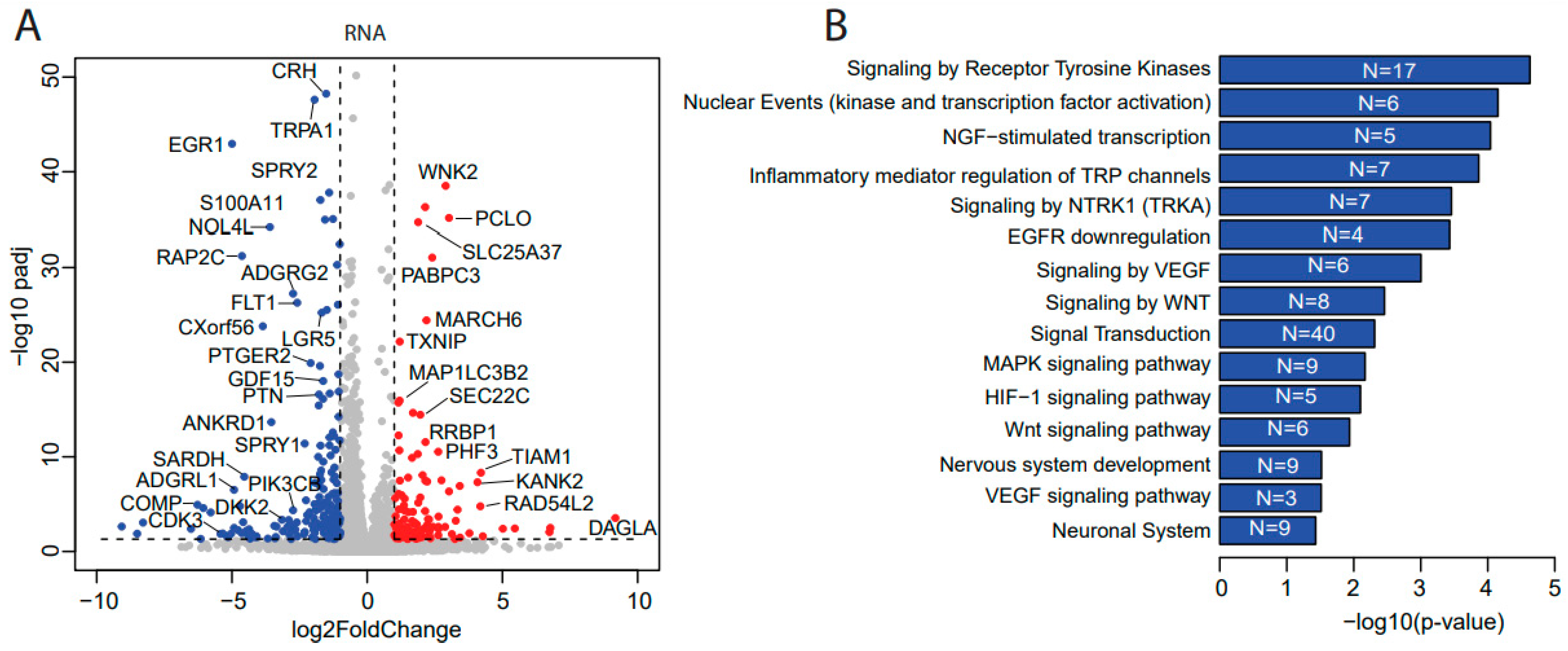
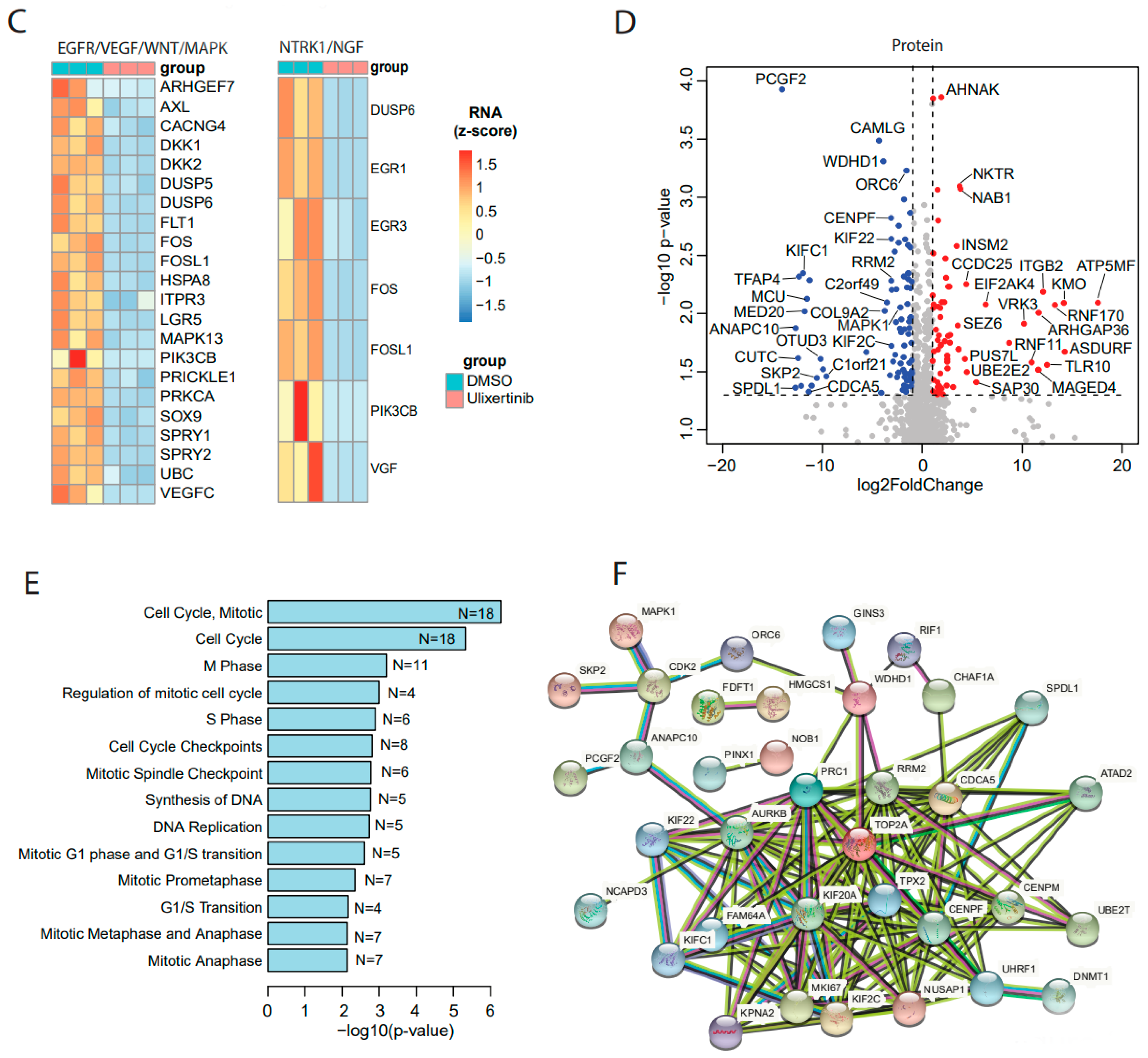
3.3. Ulixertinib Induces Extensive Transcriptomic Changes in NB
3.4. Ulixertinib Exerts Extensive Proteomic Changes in NB
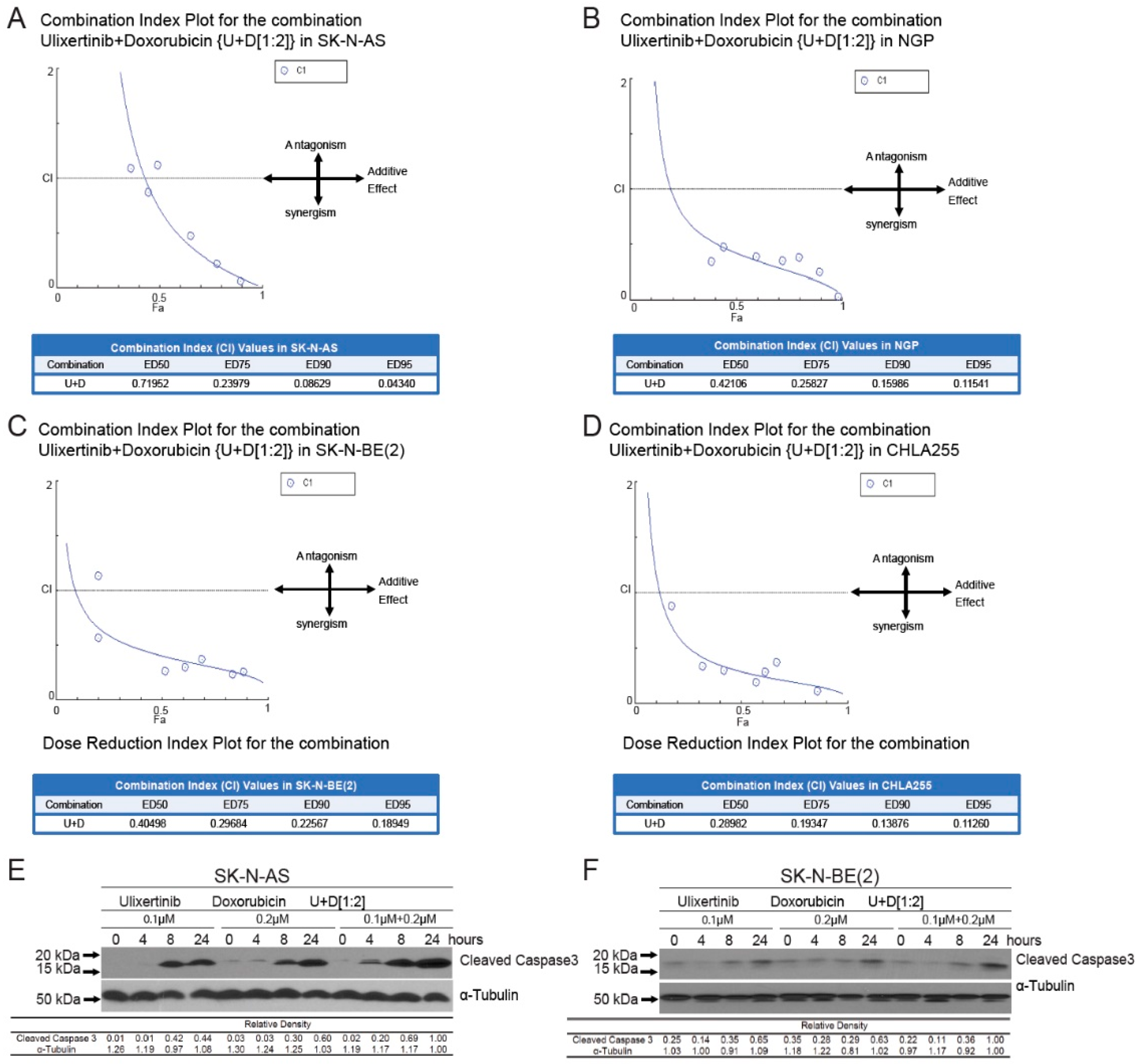
3.5. Ulixertinib Sensitizes NB to Chemotherapeutic Agent Doxorubicin
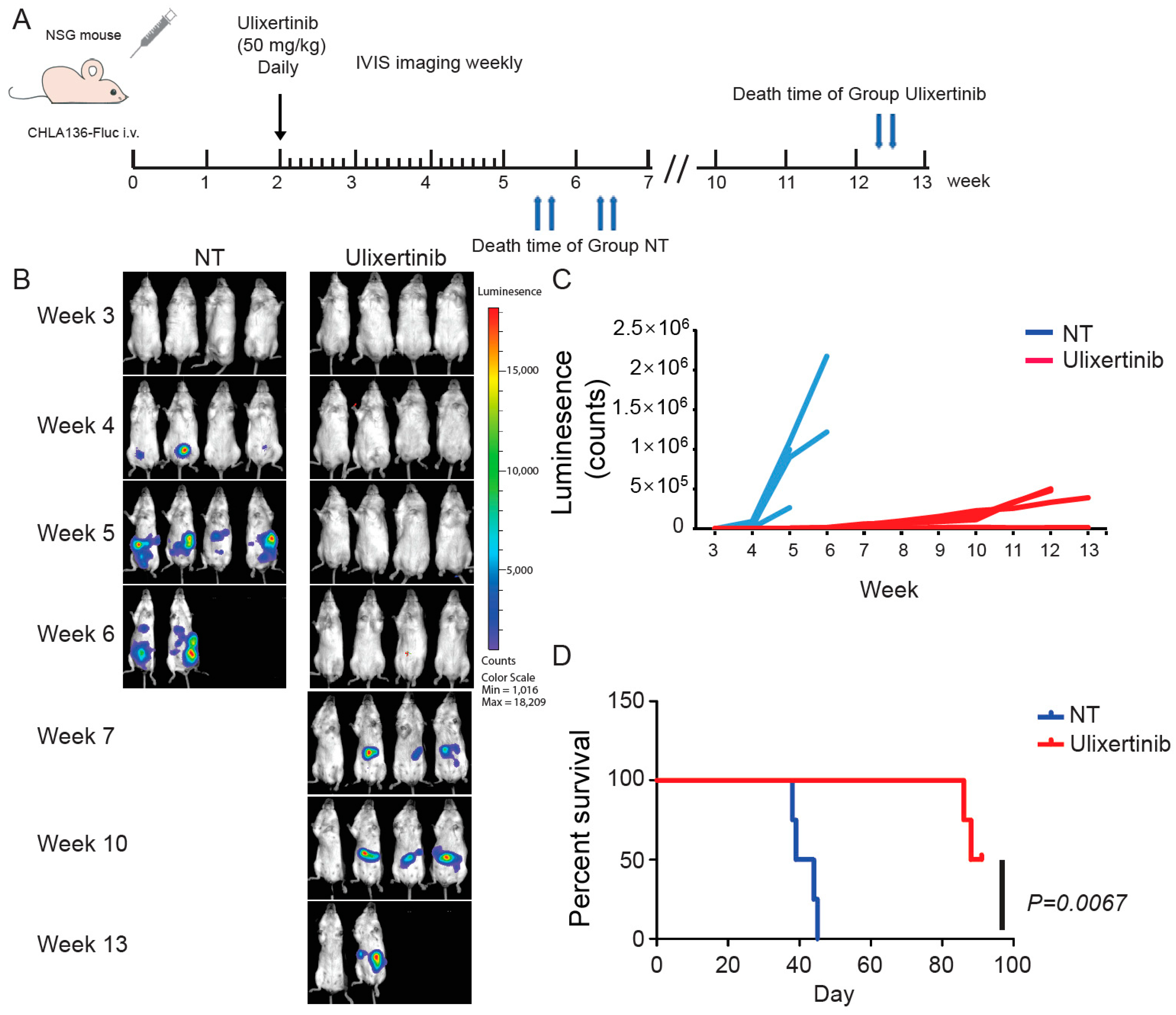
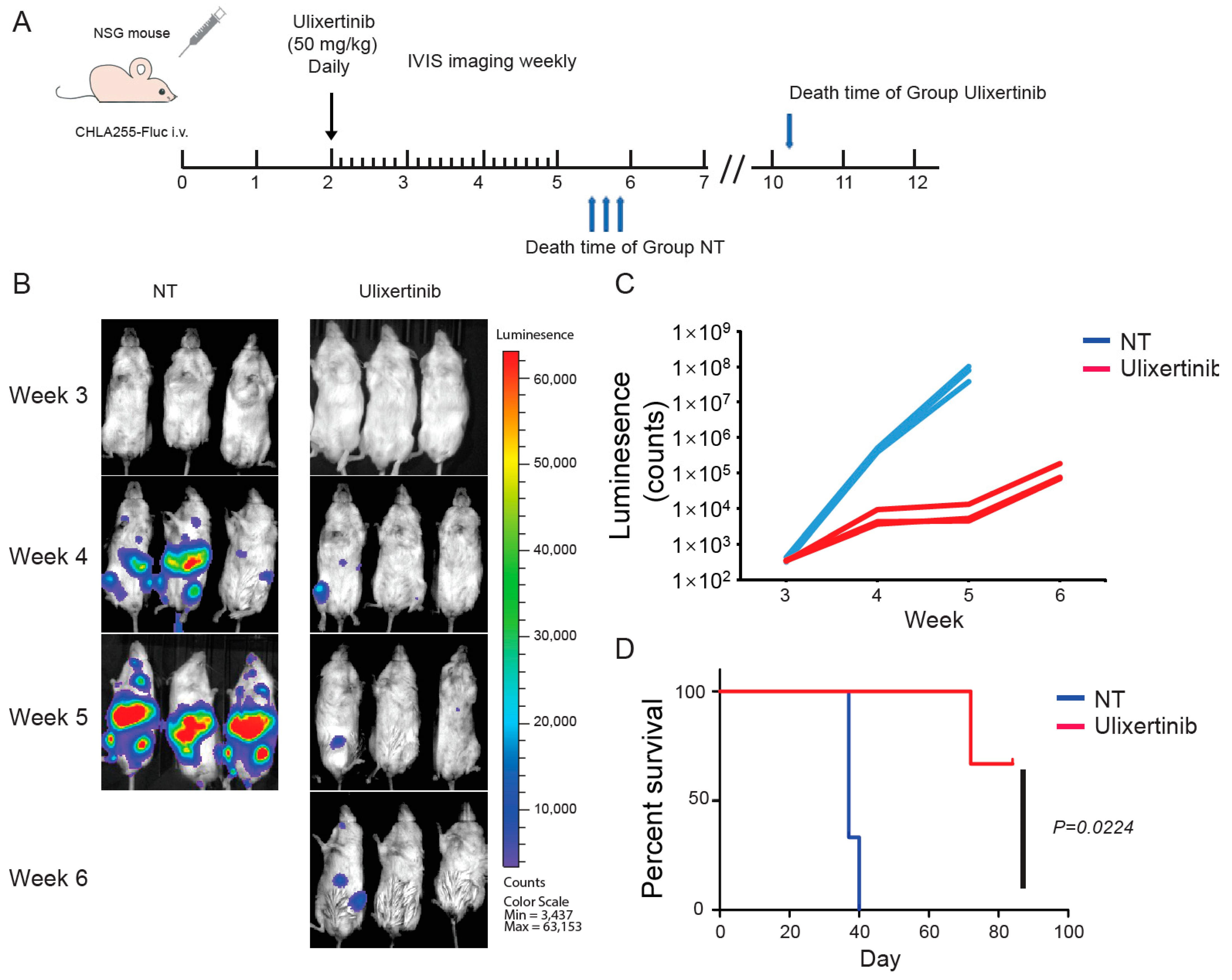
3.6. Ulixertinib Inhibits NB Tumor Growth in Xenograft Mouse Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NB | neuroblastoma |
| ERK | extracellular signal–regulated kinases |
| MAPK | mitogen-activated protein kinase |
| RAF | rapidly accelerated fibrosarcoma |
| RSK | ribosomal S6 kinase |
| MEK | mitogen-activated protein kinase |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| GSK-3β | glycogen synthase kinase 3 beta |
| SCF | Skp1-Cullin 1-F-box proteins |
| FBW7 | F-box and WD repeat domain containing 7 |
| CDK2 | cyclin dependent kinase 2 |
| ANAPC10 | anaphase promoting complex subunit 10 |
| SKP2 | S-phase kinase associated protein 2 |
| CENPM | centromere protein M |
| UBE2T | ubiquitin conjugating enzyme E2T |
| EMT | Epithelial–mesenchymal transition |
| BCL2 | B cell lymphoma 2 |
| E2F1 | E2F transcription factor 1 |
| KIFs | kinesin superfamily proteins |
| LGR5 | Leucine-rich repeat-containing G protein-coupled receptor 5 |
| RRM2 | ribonucleotide reductase regulatory subunit M2 |
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Abou-Antoun, T.; Vukmanovic, S.; Sandler, A.D. Reversible adaptive plasticity: A mechanism for neuroblastoma cell heterogeneity and chemo-resistance. Front. Oncol. 2012, 2, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizabeth, R.; Tucker, E.P. Louis Chesler, Targeting mycn and alk in resistant and relapsing neuroblastoma. Cancer Drug Resist. 2019, 2, 803–812. [Google Scholar]
- Wellbrock, C.; Karasarides, M.; Marais, R. The raf proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the erk signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent ras-mapk pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.; Chappell, W.H.; Basecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The raf/mek/erk pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Smorodinsky-Atias, K.; Soudah, N.; Engelberg, D. Mutations that confer drug-resistance, oncogenicity and intrinsic activity on the erk map kinases-current state of the art. Cells 2020, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with braf and mek inhibitors in braf(v600e) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [Green Version]
- Flemington, V.; Davies, E.J.; Robinson, D.; Sandin, L.C.; Delpuech, O.; Zhang, P.; Hanson, L.; Farrington, P.; Bell, S.; Falenta, K.; et al. Azd0364 is a potent and selective erk1/2 inhibitor that enhances antitumor activity in kras-mutant tumor models when combined with the mek inhibitor, selumetinib. Mol. Cancer Ther. 2021, 20, 238–249. [Google Scholar] [CrossRef]
- Varga, A.; Soria, J.C.; Hollebecque, A.; LoRusso, P.; Bendell, J.; Huang, S.A.; Wagle, M.C.; Okrah, K.; Liu, L.; Murray, E.; et al. A first-in-human phase i study to evaluate the erk1/2 inhibitor gdc-0994 in patients with advanced solid tumors. Clin. Cancer Res. 2020, 26, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.S.; Sim, M.Y.; Sim, H.G.; Chong, T.W.; Lau, W.K.; Cheng, C.W.; Ong, R.W.; Huynh, H. Combination of the erk inhibitor azd6244 and low-dose sorafenib in a xenograft model of human renal cell carcinoma. Int. J. Oncol. 2012, 41, 712–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Xia, W.; Liu, J.C.; Yang, J.Y.; Lee, D.F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes gsk-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Knoepfler, P.S.; Kenney, A.M. Neural precursor cycling at sonic speed: N-myc pedals, gsk-3 brakes. Cell Cycle 2006, 5, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ge, L.; Wang, M.; Carr, B.I. Phosphorylation regulates myc expression via prolonged activation of the mitogen-activated protein kinase pathway. J. Cell Physiol. 2006, 208, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Chan, H.M.; Li, Z.; Lin, C.; Nicholls, J.; Chen, C.F.; Lee, P.Y.; Lui, V.; Bacher, M.; Tam, P.K. Upregulation of macrophage migration inhibitory factor contributes to induced n-myc expression by the activation of erk signaling pathway and increased expression of interleukin-8 and vegf in neuroblastoma. Oncogene 2004, 23, 4146–4154. [Google Scholar] [CrossRef] [Green Version]
- Gogolin, S.; Dreidax, D.; Becker, G.; Ehemann, V.; Schwab, M.; Westermann, F. Mycn/myc-mediated drug resistance mechanisms in neuroblastoma. Int. J. Clin. Pharmacol. Ther. 2010, 48, 489–491. [Google Scholar] [CrossRef]
- Li, L.; Osdal, T.; Ho, Y.; Chun, S.; McDonald, T.; Agarwal, P.; Lin, A.; Chu, S.; Qi, J.; Li, L.; et al. Sirt1 activation by a c-myc oncogenic network promotes the maintenance and drug resistance of human flt3-itd acute myeloid leukemia stem cells. Cell Stem Cell 2014, 15, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, J.; Yang, X.; Fang, C.; Xu, H.; Xi, X. Sall4 as an epithelial-mesenchymal transition and drug resistance inducer through the regulation of c-myc in endometrial cancer. PLoS ONE 2015, 10, e0138515. [Google Scholar] [CrossRef]
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating beta-catenin/c-myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46. [Google Scholar] [CrossRef]
- Van Waardenburg, R.C.; Prins, J.; Meijer, C.; Uges, D.R.; De Vries, E.G.; Mulder, N.H. Effects of c-myc oncogene modulation on drug resistance in human small cell lung carcinoma cell lines. Anticancer Res. 1996, 16, 1963–1970. [Google Scholar] [PubMed]
- Parasido, E.M.; Avetian, G.S.; Brody, J.; Winter, J.; Londin, E.; Pishvaian, M.; Glasgow, E.; Byers, S.; Narla, G.; Albanese, C. Targeting c-myc and mapk pathway to overcome pancreatic cancer drug resistance. Cancer Res. 2019, 79 (Suppl. S13), 1283. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the mapk signaling pathway in cancer: Promising preclinical activity with the novel selective erk1/2 inhibitor bvd-523 (ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C.; Talaly, P. A simple generalized equation for the analysis of multiple inhibitions of michaelis-menten kinetic systems. J. Biol. Chem. 1977, 252, 6438–6442. [Google Scholar] [CrossRef]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. Nkt cells coexpressing a gd2-specific chimeric antigen receptor and il15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, B.; Muthugounder, S.; Jambon, S.; Tibbetts, R.; Hung, L.; Bassiri, H.; Hogarty, M.D.; Barrett, D.M.; Shimada, H.; Asgharzadeh, S. Preclinical assessment of the efficacy and specificity of gd2-b7h3 synnotch car-t in metastatic neuroblastoma. Nat. Commun. 2021, 12, 511. [Google Scholar] [CrossRef]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. Consensuspathdb—A database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. String v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Megison, M.L.; Gillory, L.A.; Beierle, E.A. Cell survival signaling in neuroblastoma. Anticancer Agents Med. Chem. 2013, 13, 563–575. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Mazanek, P.; Dam, V.; Wang, Q.; Zhao, H.; Guo, R.; Jagannathan, J.; Cnaan, A.; Maris, J.M.; Hogarty, M.D. Deregulated wnt/beta-catenin program in high-risk neuroblastomas without mycn amplification. Oncogene 2008, 27, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Funke, L.; Bracht, T.; Oeck, S.; Schork, K.; Stepath, M.; Dreesmann, S.; Eisenacher, M.; Sitek, B.; Schramm, A. Ntrk1/trka signaling in neuroblastoma cells induces nuclear reorganization and intra-nuclear aggregation of lamin a/c. Cancers 2021, 13, 5293. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jhuang, Y.L.; Chen, Y.L.; Jeng, Y.M.; Yuan, R.H. Paradoxical overexpression of mbnl2 in hepatocellular carcinoma inhibits tumor growth and invasion. Oncotarget 2016, 7, 65589–65601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Fernandez, M.; Kissel, H.; Brown, S.; Gorenc, T.; Schile, A.J.; Rafii, S.; Larisch, S.; Steller, H. Sept4/arts is required for stem cell apoptosis and tumor suppression. Genes Dev. 2010, 24, 2282–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterhoff, J.K.; Penninkhof, F.; Brinkmann, A.O.; Anton Grootegoed, J.; Blok, L.J. Reps2/pob1 is downregulated during human prostate cancer progression and inhibits growth factor signalling in prostate cancer cells. Oncogene 2003, 22, 2920–2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sun, Q.; Liu, K.; Zhang, L.; Lin, N.; You, K.; Liu, M.; Kon, N.; Tian, F.; Mao, Z.; et al. Otud5 cooperates with trim25 in transcriptional regulation and tumor progression via deubiquitination activity. Nat. Commun. 2020, 11, 4184. [Google Scholar] [CrossRef]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. Txnip: A double-edged sword in disease and therapeutic outlook. Oxid. Med. Cell. Longev. 2022, 2022, 7805115. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Miki, H.; Setou, M.; Kaneshiro, K.; Hirokawa, N. All kinesin superfamily protein, kif, genes in mouse and human. Proc. Natl. Acad. Sci USA 2001, 98, 7004–7011. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Takano, R.; Yokochi, T.; Akter, J.; Nakamura, Y.; Nakagawara, A.; Tatsumi, Y. Programmed expression of pro-apoptotic bmcc1 during apoptosis, triggered by DNA damage in neuroblastoma cells. BMC Cancer 2019, 19, 542. [Google Scholar] [CrossRef]
- Li, H.; Yu, Y.; Zhao, Y.; Wu, D.; Yu, X.; Lu, J.; Chen, Z.; Zhang, H.; Hu, Y.; Zhai, Y.; et al. Small molecule inhibitor agerafenib effectively suppresses neuroblastoma tumor growth in mouse models via inhibiting erk mapk signaling. Cancer Lett. 2019, 457, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Krstin, S.; Wang, S.; Wink, M. Capsaicin and piperine can overcome multidrug resistance in cancer cells to doxorubicin. Molecules 2018, 23, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, H.M.; Lai, D.K.; Falchook, G.S. Extracellular signal-regulated kinase (erk) inhibitors in oncology clinical trials. J. Immunother. Precis. Oncol. 2019, 2, 10–16. [Google Scholar] [CrossRef]
- Mendzelevski, B.; Ferber, G.; Janku, F.; Li, B.T.; Sullivan, R.J.; Welsch, D.; Chi, W.; Jackson, J.; Weng, O.; Sager, P.T. Effect of ulixertinib, a novel erk1/2 inhibitor, on the qt/qtc interval in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2018, 81, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weekes, C.; Lockhart, A.; LoRusso, P.; Murray, E.; Park, E.; Tagen, M.; Singh, J.; Sarkar, I.; Mueller, L.; Dokainish, H.; et al. A phase ib study to evaluate the mek inhibitor cobimetinib in combination with the erk1/2 inhibitor gdc-0994 in patients with advanced solid tumors. Oncologist 2020, 25, 833-e1438. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Xu, M.; Li, L.; Grierson, P.; Dodhiawala, P.; Highkin, M.; Zhang, D.; Li, Q.; Wang-Gillam, A.; Lim, K.H. Concurrent her or pi3k inhibition potentiates the antitumor effect of the erk inhibitor ulixertinib in preclinical pancreatic cancer models. Mol. Cancer Ther. 2018, 17, 2144–2155. [Google Scholar] [CrossRef] [Green Version]
- Sigaud, R.; Rosch, L.; Gatzweiler, C.; Benzel, J.; von Soosten, L.; Peterziel, H.; Selt, F.; Najafi, S.; Ayhan, S.; Gerloff, X.F.; et al. The first-in-class erk inhibitor ulixertinib shows promising activity in mapk-driven pediatric low-grade glioma models. Neuro Oncol. 2022, noac183. [Google Scholar] [CrossRef]
- Fan, X.S.; Wu, H.Y.; Yu, H.P.; Zhou, Q.; Zhang, Y.F.; Huang, Q. Expression of lgr5 in human colorectal carcinogenesis and its potential correlation with beta-catenin. Int. J. Colorectal Dis. 2010, 25, 583–590. [Google Scholar] [CrossRef]
- Tanese, K.; Fukuma, M.; Yamada, T.; Mori, T.; Yoshikawa, T.; Watanabe, W.; Ishiko, A.; Amagai, M.; Nishikawa, T.; Sakamoto, M. G-protein-coupled receptor gpr49 is up-regulated in basal cell carcinoma and promotes cell proliferation and tumor formation. Am. J. Pathol. 2008, 173, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Sakamoto, M.; Fujii, G.; Tsuiji, H.; Kenetaka, K.; Asaka, M.; Hirohashi, S. Overexpression of orphan g-protein-coupled receptor, gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology 2003, 37, 528–533. [Google Scholar] [CrossRef]
- Vieira, G.C.; Chockalingam, S.; Melegh, Z.; Greenhough, A.; Malik, S.; Szemes, M.; Park, J.H.; Kaidi, A.; Zhou, L.; Catchpoole, D.; et al. Lgr5 regulates pro-survival mek/erk and proliferative wnt/beta-catenin signalling in neuroblastoma. Oncotarget 2015, 6, 40053–40067. [Google Scholar] [CrossRef] [PubMed]
- Ciro, M.; Prosperini, E.; Quarto, M.; Grazini, U.; Walfridsson, J.; McBlane, F.; Nucifero, P.; Pacchiana, G.; Capra, M.; Christensen, J.; et al. Atad2 is a novel cofactor for myc, overexpressed and amplified in aggressive tumors. Cancer Res. 2009, 69, 8491–8498. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Lu, X.; Wang, Y.; He, H.; Meng, X.; Xia, S.; Zhen, K.; Liu, Y. Epigenetic high regulation of atad2 regulates the hh pathway in human hepatocellular carcinoma. Int. J. Oncol. 2014, 45, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct phosphorylation and stabilization of myc by aurora b kinase promote t-cell leukemogenesis. Cancer Cell 2020, 37, 200–215 e205. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sato, S.; Tsuruo, T. Phosphorylation of p27kip1 at threonine 198 by p90 ribosomal protein s6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 2003, 278, 49254–49260. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Gong, W.; Cao, J.; Li, X.; Tao, D.; Hu, J.; Gong, J. Influence of cdk1 and cdk2 sirna interference on tumor cell cycle and cell apoptosis. Chin-Ger. J. Clin. Oncol. 2009, 8, 371–374. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Chattopadhyay, S.; Siddiqui, F.A.; Ur Rehman, A.; Siddiqui, S.; Prakasam, G.; Khan, A.; Sultana, S.; Bamezai, R.N. Silibinin induces metabolic crisis in triple-negative breast cancer cells by modulating egfr-myc-txnip axis: Potential therapeutic implications. FEBS J. 2021, 288, 471–485. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Potter, J.J.; Koganti, L.; Meltzer, S.J.; Mezey, E. Effects of vitamin d3 stimulation of thioredoxin-interacting protein in hepatocellular carcinoma. Hepatol. Res. 2014, 44, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.Y.; Yu, F.X.; Luo, Y.; Hagen, T. Oncogenic activation of the pi3k/akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell Signal. 2016, 28, 377–383. [Google Scholar] [CrossRef]
- Jiao, D.; Huan, Y.; Zheng, J.; Wei, M.; Zheng, G.; Han, D.; Wu, J.; Xi, W.; Wei, F.; Yang, A.G.; et al. Uhrf1 promotes renal cell carcinoma progression through epigenetic regulation of txnip. Oncogene 2019, 38, 5686–5699. [Google Scholar] [CrossRef]
- Pan, X.N.; Chen, J.J.; Wang, L.X.; Xiao, R.Z.; Liu, L.L.; Fang, Z.G.; Liu, Q.; Long, Z.J.; Lin, D.J. Inhibition of c-myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PLoS ONE 2014, 9, e105381. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Kadomatsu, K.; Sato, S.; Kohno, K.; Takano, H.; Akazawa, K.; Nose, Y.; Kuwano, M. Inverse correlation between expression of multidrug resistance gene and n-myc oncogene in human neuroblastomas. Cancer Res. 1990, 50, 3043–3047. [Google Scholar] [PubMed]
- Li, Y.; Zhuo, B.; Yin, Y.; Han, T.; Li, S.; Li, Z.; Wang, J. Anti-cancer effect of oncolytic adenovirus-armed shrna targeting mycn gene on doxorubicin-resistant neuroblastoma cells. Biochem. Biophys. Res. Commun. 2017, 491, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, W.; Zhou, J.; Zhang, J.; Huang, Y.; Wang, Z. Erk inhibitor enhances everolimus efficacy through the attenuation of dntp pools in renal cell carcinoma. Mol. Ther. Nucleic Acids 2019, 14, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Amin, A.R.; Wang, D.; Koenig, L.; Nannapaneni, S.; Chen, Z.; Wang, Z.; Sica, G.; Deng, X.; Chen, Z.G.; et al. Rrm2 regulates bcl-2 in head and neck and lung cancers: A potential target for cancer therapy. Clin. Cancer Res. 2013, 19, 3416–3428. [Google Scholar] [CrossRef] [Green Version]
- Mazzu, Y.Z.; Armenia, J.; Chakraborty, G.; Yoshikawa, Y.; Coggins, S.A.; Nandakumar, S.; Gerke, T.A.; Pomerantz, M.M.; Qiu, X.; Zhao, H.; et al. A novel mechanism driving poor-prognosis prostate cancer: Overexpression of the DNA repair gene, ribonucleotide reductase small subunit m2 (rrm2). Clin. Cancer Res. 2019, 25, 4480–4492. [Google Scholar] [CrossRef] [Green Version]
- Machida, T.; Fujita, T.; Ooo, M.L.; Ohira, M.; Isogai, E.; Mihara, M.; Hirato, J.; Tomotsune, D.; Hirata, T.; Fujimori, M.; et al. Increased expression of proapoptotic bmcc1, a novel gene with the bnip2 and cdc42gap homology (bch) domain, is associated with favorable prognosis in human neuroblastomas. Oncogene 2006, 25, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, Y.; Takano, R.; Islam, M.S.; Yokochi, T.; Itami, M.; Nakamura, Y.; Nakagawara, A. Bmcc1, which is an interacting partner of bcl2, attenuates akt activity, accompanied by apoptosis. Cell Death Dis. 2015, 6, e1607. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, T.; Vagapova, E.; Spirin, P.; Rubtsov, P.; Astashkova, O.; Mikheeva, A.; Sorokin, M.; Vladimirova, U.; Suntsova, M.; Konovalov, D.; et al. Growth factor signaling predicts therapy resistance mechanisms and defines neuroblastoma subtypes. Oncogene 2021, 40, 6258–6272. [Google Scholar] [CrossRef]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. Nras status determines sensitivity to shp2 inhibitor combination therapies targeting the ras-mapk pathway in neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Zhao, Y.; Choi, J.; Shi, Z.; Guo, L.; Elizarraras, J.; Gu, A.; Cheng, F.; Pei, Y.; Lu, D.; et al. ERK Inhibitor Ulixertinib Inhibits High-Risk Neuroblastoma Growth In Vitro and In Vivo. Cancers 2022, 14, 5534. https://doi.org/10.3390/cancers14225534
Yu Y, Zhao Y, Choi J, Shi Z, Guo L, Elizarraras J, Gu A, Cheng F, Pei Y, Lu D, et al. ERK Inhibitor Ulixertinib Inhibits High-Risk Neuroblastoma Growth In Vitro and In Vivo. Cancers. 2022; 14(22):5534. https://doi.org/10.3390/cancers14225534
Chicago/Turabian StyleYu, Yang, Yanling Zhao, Jongmin Choi, Zhongcheng Shi, Linjie Guo, John Elizarraras, Andy Gu, Feng Cheng, Yanxin Pei, Dai Lu, and et al. 2022. "ERK Inhibitor Ulixertinib Inhibits High-Risk Neuroblastoma Growth In Vitro and In Vivo" Cancers 14, no. 22: 5534. https://doi.org/10.3390/cancers14225534