
RET Proto-Oncogene—Not Such an Obvious Starting Point in Cancer Therapy

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
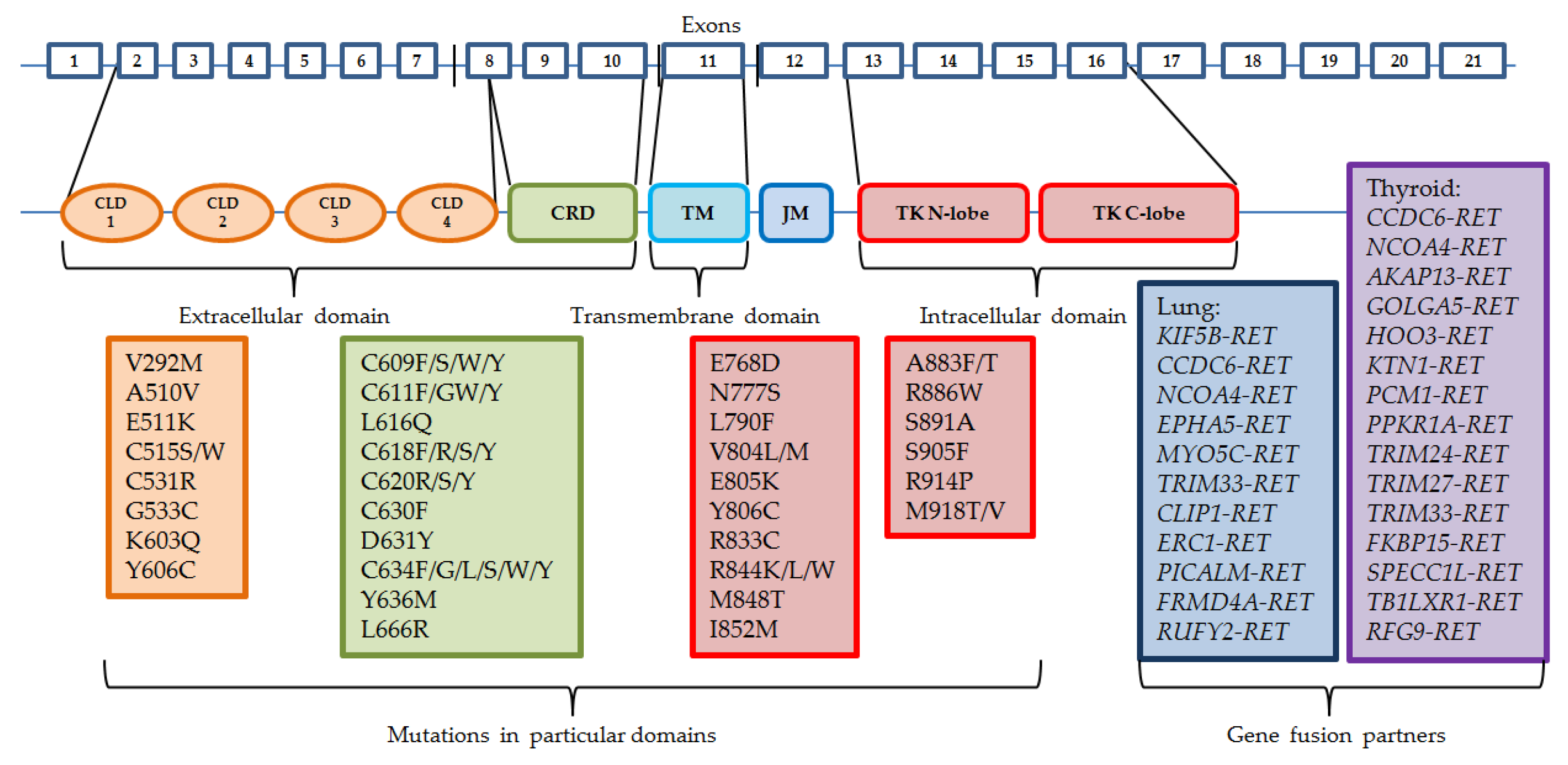
2. RET Protein and Activity
3. RET Rearrangements
4. Mutations
5. Detection of RET Alterations
6. Immunotherapy in Tumors with Genetic Changes of RET Gene
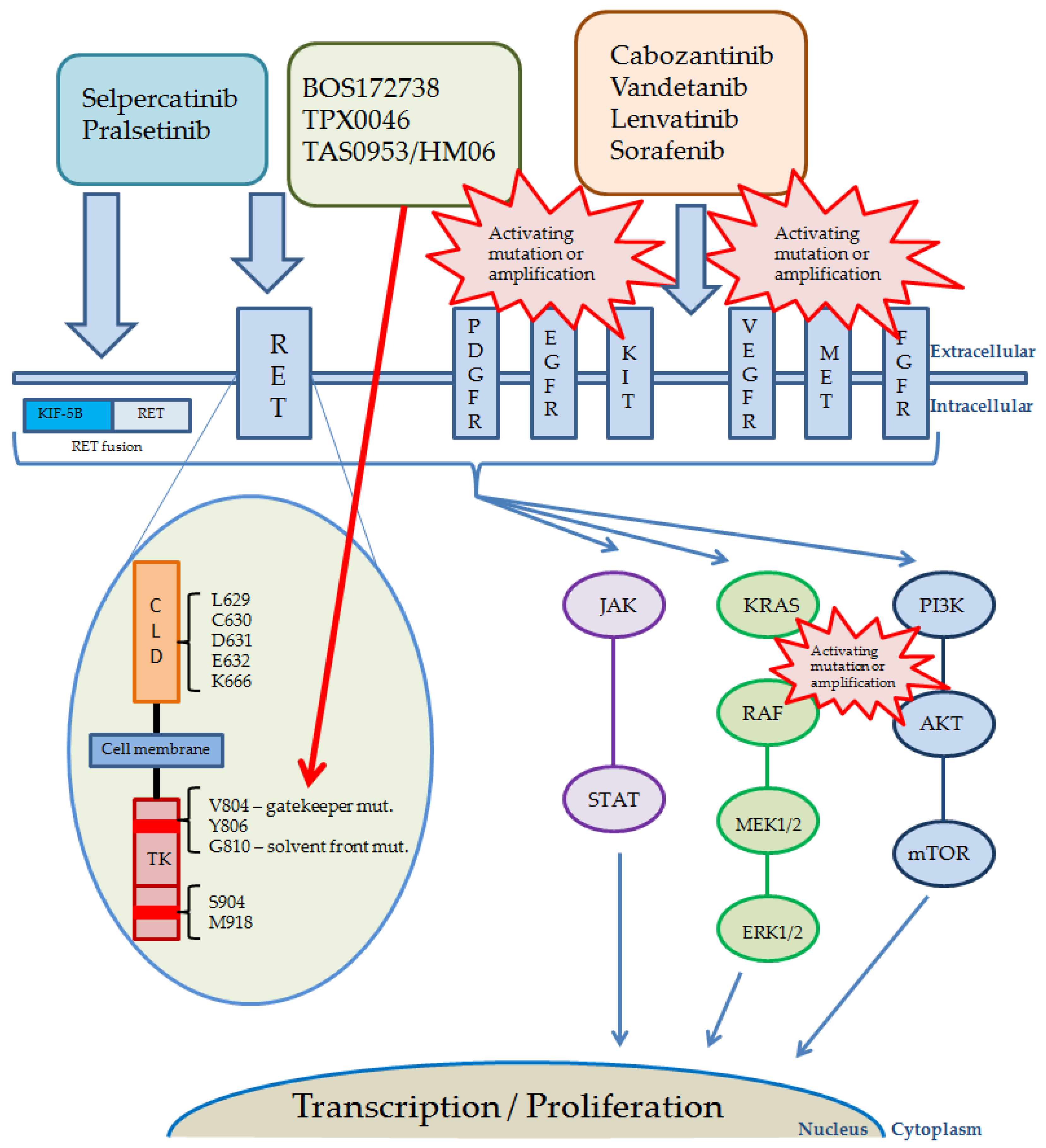
7. Treatment of RET-Altered Cancers with Multiple Kinase Inhibitors
8. RET-Specific Inhibitors
9. Next Generation Selective RET Inhibitors
10. Resistance to RET Selective Inhibitors
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine Kinase Inhibitors for Solid Tumors in the Past 20 Years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. RET Receptor Signaling: Function in Development, Metabolic Disease, and Cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2022, 98, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET Tyrosine Kinase Signaling in Development and Cancer. Cytokine Growth Factor Rev. 2005, 16, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Subbiah, V.; Marchlik, E.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1988–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccherini, I.; Bocciardi, R.; Luo, Y.; Pasini, B.; Hofstra, R.; Takahashi, M.; Romeo, G. Exon Structure and Flanking Intronic Sequences of the Human RET Proto-Oncogene. Biochem. Biophys. Res. Commun. 1993, 196, 1288–1295. [Google Scholar] [CrossRef]
- Rossel, M.; Pasini, A.; Chappuis, S.; Geneste, O.; Fournier, L.; Schuffenecker, I.; Takahashi, M.; van Grunsven, L.A.; Urdiales, J.L.; Rudkin, B.B.; et al. Distinct Biological Properties of Two RET Isoforms Activated by MEN 2A and MEN 2B Mutations. Oncogene 1997, 14, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Le Hir, H.; Charlet-Berguerand, N.; Gimenez-Roqueplo, A.; Mannelli, M.; Plouin, P.; de Franciscis, V.; Thermes, C. Relative Expression of the RET9 and RET51 Isoforms in Human Pheochromocytomas. Oncology 2000, 58, 311–318. [Google Scholar] [CrossRef]
- Lian, E.Y.; Maritan, S.M.; Cockburn, J.G.; Kasaian, K.; Crupi, M.J.F.; Hurlbut, D.; Jones, S.J.M.; Wiseman, S.M.; Mulligan, L.M. Differential Roles of RET Isoforms in Medullary and Papillary Thyroid Carcinomas. Endocr. Relat. Cancer 2017, 24, 53–69. [Google Scholar] [CrossRef] [Green Version]
- Anders, J.; Kjar, S.; Ibáñez, C.F. Molecular Modeling of the Extracellular Domain of the RET Receptor Tyrosine Kinase Reveals Multiple Cadherin-like Domains and a Calcium-Binding Site. J. Biol. Chem. 2001, 276, 35808–35817. [Google Scholar] [CrossRef]
- Liu, X.; Vega, Q.C.; Decker, R.A.; Pandey, A.; Worby, C.A.; Dixon, J.E. Oncogenic RET Receptors Display Different Autophosphorylation Sites and Substrate Binding Specificities. J. Biol. Chem. 1996, 271, 5309–5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez, C.F. Structure and Physiology of the RET Receptor Tyrosine Kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a009134. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-Driven Cancers: Lessons from Evolving Preclinical and Clinical Landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, C.; Poudel, P.P.; Ghosh, A.; Kalthur, S.G. The RET Gene Encodes RET Protein, Which Triggers Intracellular Signaling Pathways for Enteric Neurogenesis, and RET Mutation Results in Hirschsprung’s Disease. AIMS Neurosci. 2022, 9, 128–149. [Google Scholar] [CrossRef]
- Mizukami, T.; Shiraishi, K.; Shimada, Y.; Ogiwara, H.; Tsuta, K.; Ichikawa, H.; Sakamoto, H.; Kato, M.; Shibata, T.; Nakano, T.; et al. Molecular Mechanisms Underlying Oncogenic RET Fusion in Lung Adenocarcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2014, 9, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Stringer, J.R.; Blough, R.; Medvedovic, M.; Fagin, J.A.; Nikiforov, Y.E. Proximity of Chromosomal Loci That Participate in Radiation-Induced Rearrangements in Human Cells. Science 2000, 290, 138–141. [Google Scholar] [CrossRef]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Na, J.M.; Lee, G.-W.; Lee, S.J.; Kim, J.D.; Yang, J.W. EGFR-Mutated Pulmonary Adenocarcinoma with Concurrent PIK3CA Mutation, and with Acquired RET Fusion and EGFR T790M Mutation after Afatinib Therapy. J. Pathol. Transl. Med. 2021, 55, 79–82. [Google Scholar] [CrossRef]
- Williams, D. Radiation Carcinogenesis: Lessons from Chernobyl. Oncogene 2008, 27 (Suppl. 2), S9–S18. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Vorontsova, T.; Cosci, B.; Veremeychik, V.; Kuchinskaya, E.; Basolo, F.; Demidchik, E.P.; Miccoli, P.; Pinchera, A.; et al. RET/PTC Rearrangements in Thyroid Nodules: Studies in Irradiated and Not Irradiated, Malignant and Benign Thyroid Lesions in Children and Adults. J. Clin. Endocrinol. Metab. 2001, 86, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- Hamatani, K.; Eguchi, H.; Ito, R.; Mukai, M.; Takahashi, K.; Taga, M.; Imai, K.; Cologne, J.; Soda, M.; Arihiro, K.; et al. RET/PTC Rearrangements Preferentially Occurred in Papillary Thyroid Cancer among Atomic Bomb Survivors Exposed to High Radiation Dose. Cancer Res. 2008, 68, 7176–7182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Seyama, T.; Iwamoto, K.S.; Hayashi, T.; Mizuno, T.; Tsuyama, N.; Dohi, K.; Nakamura, N.; Akiyama, M. In Vitro Irradiation Is Able to Cause RET Oncogene Rearrangement. Cancer Res. 1993, 53, 2940–2943. [Google Scholar] [PubMed]
- Fugazzola, L.; Pilotti, S.; Pinchera, A.; Vorontsova, T.V.; Mondellini, P.; Bongarzone, I.; Greco, A.; Astakhova, L.; Butti, M.G.; Demidchik, E.P. Oncogenic Rearrangements of the RET Proto-Oncogene in Papillary Thyroid Carcinomas from Children Exposed to the Chernobyl Nuclear Accident. Cancer Res. 1995, 55, 5617–5620. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Romei, C.; Ciampi, R.; Elisei, R. A Comprehensive Overview of the Role of the RET Proto-Oncogene in Thyroid Carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes 2020, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.S.; Gujral, T.S.; Peng, S.; Asa, S.L.; Mulligan, L.M. Transcript Level Modulates the Inherent Oncogenicity of RET/PTC Oncoproteins. Cancer Res. 2009, 69, 4861–4869. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET Fusions Define a Unique Molecular and Clinicopathologic Subtype of Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef]
- Kazdal, D.; Hofman, V.; Christopoulos, P.; Ilié, M.; Stenzinger, A.; Hofman, P. Fusion-Positive Non-Small Cell Lung Carcinoma: Biological Principles, Clinical Practice, and Diagnostic Implications. Genes. Chromosomes Cancer 2022, 61, 244–260. [Google Scholar] [CrossRef]
- Belli, C.; Penault-Llorca, F.; Ladanyi, M.; Normanno, N.; Scoazec, J.-Y.; Lacroix, L.; Reis-Filho, J.S.; Subbiah, V.; Gainor, J.F.; Endris, V.; et al. ESMO Recommendations on the Standard Methods to Detect RET Fusions and Mutations in Daily Practice and Clinical Research. Ann. Oncol. 2021, 32, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Chao, B.H.; Briesewitz, R.; Villalona-Calero, M.A. RET Fusion Genes in Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4439–4441. [Google Scholar] [CrossRef] [PubMed]
- Skálová, A.; Vanecek, T.; Uro-Coste, E.; Bishop, J.A.; Weinreb, I.; Thompson, L.D.R.; de Sanctis, S.; Schiavo-Lena, M.; Laco, J.; Badoual, C.; et al. Molecular Profiling of Salivary Gland Intraductal Carcinoma Revealed a Subset of Tumors Harboring NCOA4-RET and Novel TRIM27-RET Fusions: A Report of 17 Cases. Am. J. Surg. Pathol. 2018, 42, 1445–1455. [Google Scholar] [CrossRef]
- Santos, C.; Sanz-Pamplona, R.; Salazar, R. RET-Fusions: A Novel Paradigm in Colorectal Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Paratala, B.S.; Chung, J.H.; Williams, C.B.; Yilmazel, B.; Petrosky, W.; Williams, K.; Schrock, A.B.; Gay, L.M.; Lee, E.; Dolfi, S.C.; et al. RET Rearrangements Are Actionable Alterations in Breast Cancer. Nat. Commun. 2018, 9, 4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase Fusions Are Frequent in Spitz Tumours and Spitzoid Melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.-F.; et al. RET Fusion Genes Are Associated with Chronic Myelomonocytic Leukemia and Enhance Monocytic Differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Raue, F.; Frank-Raue, K. Update on Multiple Endocrine Neoplasia Type 2: Focus on Medullary Thyroid Carcinoma. J. Endocr. Soc. 2018, 2, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Krampitz, G.W.; Norton, J.A. RET Gene Mutations (Genotype and Phenotype) of Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma. Cancer 2014, 120, 1920–1931. [Google Scholar] [CrossRef]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A. Mutations in the RET Proto-Oncogene Are Associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993, 2, 851–856. [Google Scholar] [CrossRef]
- Hofstra, R.M.; Landsvater, R.M.; Ceccherini, I.; Stulp, R.P.; Stelwagen, T.; Luo, Y.; Pasini, B.; Höppener, J.W.; van Amstel, H.K.; Romeo, G. A Mutation in the RET Proto-Oncogene Associated with Multiple Endocrine Neoplasia Type 2B and Sporadic Medullary Thyroid Carcinoma. Nature 1994, 367, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Román-Gil, M.S.; Pozas, J.; Rosero-Rodríguez, D.; Chamorro-Pérez, J.; Ruiz-Granados, Á.; Caracuel, I.R.; Grande, E.; Molina-Cerrillo, J.; Alonso-Gordoa, T. Resistance to RET Targeted Therapy in Thyroid Cancer: Molecular Basis and Overcoming Strategies. Cancer Treat. Rev. 2022, 105, 102372. [Google Scholar] [CrossRef] [PubMed]
- Asai, N.; Iwashita, T.; Matsuyama, M.; Takahashi, M. Mechanism of Activation of the Ret Proto-Oncogene by Multiple Endocrine Neoplasia 2A Mutations. Mol. Cell. Biol. 1995, 15, 1613–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.; Carlomagno, F.; Romano, A.; Bottaro, D.P.; Dathan, N.A.; Grieco, M.; Fusco, A.; Vecchio, G.; Matoskova, B.; Kraus, M.H. Activation of RET as a Dominant Transforming Gene by Germline Mutations of MEN2A and MEN2B. Science 1995, 267, 381–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, C.; Anand, S.; Gainor, J.F.; Penault-Llorca, F.; Subbiah, V.; Drilon, A.; Andrè, F.; Curigliano, G. Progresses Toward Precision Medicine in RET-Altered Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 6102–6111. [Google Scholar] [CrossRef]
- Thein, K.Z.; Velcheti, V.; Mooers, B.H.M.; Wu, J.; Subbiah, V. Precision Therapy for RET-Altered Cancers with RET Inhibitors. Trends Cancer 2021, 7, 1074–1088. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET Revisited: Expanding the Oncogenic Portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Yang, Y.-Z.; Hu, W.-M.; Xia, L.-P.; He, W.-Z. Association between Somatic RET Mutations and Clinical and Genetic Characteristics in Patients with Metastatic Colorectal Cancer. Cancer Med. 2021, 10, 8876–8882. [Google Scholar] [CrossRef]
- Edery, P.; Lyonnet, S.; Mulligan, L.M.; Pelet, A.; Dow, E.; Abel, L.; Holder, S.; Nihoul-Fékété, C.; Ponder, B.A.; Munnich, A. Mutations of the RET Proto-Oncogene in Hirschsprung’s Disease. Nature 1994, 367, 378–380. [Google Scholar] [CrossRef]
- Angrist, M.; Bolk, S.; Thiel, B.; Puffenberger, E.G.; Hofstra, R.M.; Buys, C.H.; Cass, D.T.; Chakravarti, A. Mutation Analysis of the RET Receptor Tyrosine Kinase in Hirschsprung Disease. Hum. Mol. Genet. 1995, 4, 821–830. [Google Scholar] [CrossRef]
- Sijmons, R.H.; Hofstra, R.M.; Wijburg, F.A.; Links, T.P.; Zwierstra, R.P.; Vermey, A.; Aronson, D.C.; Tan-Sindhunata, G.; Brouwers-Smalbraak, G.J.; Maas, S.M.; et al. Oncological Implications of RET Gene Mutations in Hirschsprung’s Disease. Gut 1998, 43, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; De Vita, G.; Berlingieri, M.; de Franciscis, V.; Melillo, R.; Colantuoni, V.; Kraus, M.; Di Fiore, P.P.; Fusco, A.; Santoro, M. Molecular Heterogeneity of RET Loss of Function in Hirschsprung’s Disease. EMBO J. 1996, 15, 2717–2725. [Google Scholar] [CrossRef]
- Takahashi, M.; Kawai, K.; Asai, N. Roles of the RET Proto-Oncogene in Cancer and Development. JMA J. 2020, 3, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-R.; Aypar, U.; Rosen, E.Y.; Mata, D.A.; Benayed, R.; Mullaney, K.; Jayakumaran, G.; Zhang, Y.; Frosina, D.; Drilon, A.; et al. A Performance Comparison of Commonly Used Assays to Detect RET Fusions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Clark, D.P.; Hawkins, A.L.; Morsberger, L.A.; Griffin, C.A. A Break-Apart Fluorescence in Situ Hybridization Assay for Detecting RET Translocations in Papillary Thyroid Carcinoma. Cancer Genet. Cytogenet. 2007, 178, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of Cell-Free DNA from 32,989 Advanced Cancers Reveals Novel Co-Occurring Activating RET Alterations and Oncogenic Signaling Pathway Aberrations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini-Spaltro, A.; Farnedi, A.; Calistri, D.; Rengucci, C.; Prisinzano, G.; Chiadini, E.; Capelli, L.; Angeli, D.; Bennati, C.; Valli, M.; et al. The Role of Next-Generation Sequencing in Detecting Gene Fusions with Known and Unknown Partners: A Single-Center Experience with Methodologies’ Integration. Hum. Pathol. 2022, 123, 20–30. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Nagasaka, M.; Zhu, V.W. Liquid Biopsy to Identify Actionable Genomic Alterations. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2018, 38, 978–997. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.; Andreev-Drakhlin, A.Y.; Roszik, J.; Huang, L.; Liu, S.; Hess, K.; Cabanillas, M.; Hu, M.I.; Busaidy, N.L.; Sherman, S.I.; et al. Responsiveness to Immune Checkpoint Inhibitors versus Other Systemic Therapies in RET-Aberrant Malignancies. ESMO Open 2020, 5, e000799. [Google Scholar] [CrossRef]
- Offin, M.; Guo, R.; Wu, S.L.; Sabari, J.; Land, J.D.; Ni, A.; Montecalvo, J.; Halpenny, D.F.; Buie, L.W.; Pak, T.; et al. Immunophenotype and Response to Immunotherapy of RET-Rearranged Lung Cancers. JCO Precis. Oncol. 2019, 3, PO.18.00386. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ku, B.M.; Shim, J.H.; La Choi, Y.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. Characteristics and Outcomes of RET-Rearranged Korean Non-Small Cell Lung Cancer Patients in Real-World Practice. Jpn. J. Clin. Oncol. 2020, 50, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Knetki-Wróblewska, M.; Wojas-Krawczyk, K.; Kowalski, D.M.; Krzakowski, M. Non-Small-Cell Lung Cancer Patients with Coexistence of High PD-L1 Expression and RET Fusion-Which Path Should We Follow? Case Reports and Literature Review. J. Clin. Med. 2022, 11, 1630. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Lin, J.J.; Filleron, T.; Ni, A.; Milia, J.; Bergagnini, I.; Hatzoglou, V.; Velcheti, V.; Offin, M.; Li, B.; et al. Frequency of Brain Metastases and Multikinase Inhibitor Outcomes in Patients With RET-Rearranged Lung Cancers. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 1595–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in Patients with Previously Treated RET-Rearranged Advanced Non-Small-Cell Lung Cancer (LURET): An Open-Label, Multicentre Phase 2 Trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Hida, T.; Velcheti, V.; Reckamp, K.L.; Nokihara, H.; Sachdev, P.; Kubota, T.; Nakada, T.; Dutcus, C.E.; Ren, M.; Tamura, T. A Phase 2 Study of Lenvatinib in Patients with RET Fusion-Positive Lung Adenocarcinoma. Lung Cancer Amst. Neth. 2019, 138, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet Lond. Engl. 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jin, M.; Jeon, M.J.; Kim, E.Y.; Shin, D.Y.; Lim, D.J.; Kim, B.H.; Kang, H.-C.; Kim, W.B.; Shong, Y.K.; et al. Lenvatinib Compared with Sorafenib as a First-Line Treatment for Radioactive Iodine-Refractory, Progressive, Differentiated Thyroid Carcinoma: Real-World Outcomes in a Multicenter Retrospective Cohort Study. Thyroid Off. J. Am. Thyroid Assoc. 2022. [Google Scholar] [CrossRef]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Elisei, R.; Müller, S.; Schöffski, P.; Brose, M.; Shah, M.; Licitra, L.; Krajewska, J.; Kreissl, M.C.; Niederle, B.; et al. Overall Survival Analysis of EXAM, a Phase III Trial of Cabozantinib in Patients with Radiographically Progressive Medullary Thyroid Carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Matrone, A.; Gambale, C.; Prete, A.; Elisei, R. Sporadic Medullary Thyroid Carcinoma: Towards a Precision Medicine. Front. Endocrinol. 2022, 13, 864253. [Google Scholar] [CrossRef] [PubMed]
- Li, G.G.; Somwar, R.; Joseph, J.; Smith, R.S.; Hayashi, T.; Martin, L.; Franovic, A.; Schairer, A.; Martin, E.; Riely, G.J.; et al. Antitumor Activity of RXDX-105 in Multiple Cancer Types with RET Rearrangements or Mutations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2981–2990. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Fu, S.; Patel, M.R.; Fakih, M.; Wang, D.; Olszanski, A.J.; Morgensztern, D.; Liu, S.V.; Cho, B.C.; Bazhenova, L.; et al. A Phase I/Ib Trial of the VEGFR-Sparing Multikinase RET Inhibitor RXDX-105. Cancer Discov. 2019, 9, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Bradford, D.; Larkins, E.; Mushti, S.L.; Rodriguez, L.; Skinner, A.M.; Helms, W.S.; Price, L.S.L.; Zirkelbach, J.F.; Li, Y.; Liu, J.; et al. FDA Approval Summary: Selpercatinib for the Treatment of Lung and Thyroid Cancers with RET Gene Mutations or Fusions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; et al. FDA Approval Summary: Pralsetinib for the Treatment of Lung and Thyroid Cancers With RET Gene Mutations or Fusions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- EMA Meeting Highlights from the Committee for Medicinal Products Human Use (CHMP) 7–10 December 2020. Available online: https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-7-10-december-2020 (accessed on 4 August 2022).
- EMA Meeting Highlights from the Committee for Medicinal Products Human Use (CHMP) 13–16 September 2021. Available online: https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-13-16-september-2021 (accessed on 4 August 2022).
- Della Corte, C.M.; Morgillo, F. Rethinking Treatment for RET-Altered Lung and Thyroid Cancers: Selpercatinib Approval by the EMA. ESMO Open 2021, 6, 100041. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Curigliano, G.; Kim, D.-W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET Fusion-Positive Non-Small-Cell Lung Cancer (ARROW): A Multi-Cohort, Open-Label, Phase 1/2 Study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef]
- Curigliano, G.; Gainor, J.F.; Griesinger, F.; Thomas, M.; Subbiah, V.; Baik, C.S.; Tan, D.S.-W.; Lee, D.H.; Misch, D.; Garralda, E.; et al. Safety and Efficacy of Pralsetinib in Patients with Advanced RET Fusion-Positive Non-Small Cell Lung Cancer: Update from the ARROW Trial. J. Clin. Oncol. 2021, 39, 9089. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for Patients with Advanced or Metastatic RET-Altered Thyroid Cancer (ARROW): A Multi-Cohort, Open-Label, Registrational, Phase 1/2 Study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.-N.; Gainor, J.F.; Mansfield, A.S.; Alonso, G.; Taylor, M.H.; Zhu, V.W.; Garrido Lopez, P.; Amatu, A.; Doebele, R.C.; et al. Clinical Activity of the RET Inhibitor Pralsetinib (BLU-667) in Patients with RET Fusion+ Solid Tumors. J. Clin. Oncol. 2020, 38, 109. [Google Scholar] [CrossRef]
- Lee, D.H.; Subbiah, V.; Gainor, J.F.; Taylor, M.H.; Zhu, V.W.; Doebele, R.C.; Lopes, G.; Baik, C.; Garralda, E.; Gadgeel, S.M.; et al. Treatment with Pralsetinib (Formerly BLU-667), a Potent and Selective RET Inhibitor, Provides Rapid Clearance of CtDNA in Patients with RET-Altered Non-Small Cell Lung Cancer (NSCLC) and Medullary Thyroid Cancer (MTC). Ann. Oncol. 2019, 30, ix122. [Google Scholar] [CrossRef]
- Besse, B.; Felip, E.; Clifford, C.; Louie-Gao, M.; Green, J.; Turner, C.D.; Popat, S. AcceleRET Lung: A Phase III Study of First-Line Pralsetinib in Patients (Pts) with RET-Fusion+ Advanced/Metastatic Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2020, 38, TPS9633. [Google Scholar] [CrossRef]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural Basis of Acquired Resistance to Selpercatinib and Pralsetinib Mediated by Non-Gatekeeper RET Mutations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Hernando, J.; Tarasova, V.; Hu, M.I.; Sherman, E.J.; Brose, M.S.; Robinson, B.; Tahara, M.; Wirth, L.J.; Sashegyi, A.; Soldatenkova, V.; et al. 1927TiP LIBRETTO-531: Selpercatinib in Patients with Treatment (Tx)-Naïve RET-Mutant Medullary Thyroid Cancer (MTC). Ann. Oncol. 2020, 31, S1091. [Google Scholar] [CrossRef]
- Loong, H.H.F.; Goto, K.; Elamin, Y.Y.; Solomon, B.; Santini, F.C.; Soldatenkova, V.; Sashegyi, A.; Lin, A.B.; Lin, B.K.; Wolf, J.; et al. 1413TiP LIBRETTO-431: Selpercatinib in Treatment (Tx)-Naïve Patients with RET Fusion-Positive (RET+) Non-Small Cell Lung Cancer (NSCLC). Ann. Oncol. 2020, 31, S893. [Google Scholar] [CrossRef]
- Ortiz, M.V.; Gerdemann, U.; Raju, S.G.; Henry, D.; Smith, S.; Rothenberg, S.M.; Cox, M.C.; Proust, S.; Bender, J.G.; Frazier, A.L.; et al. Activity of the Highly Specific RET Inhibitor Selpercatinib (LOXO-292) in Pediatric Patients with Tumors Harboring RET Gene Alterations. JCO Precis. Oncol. 2020, 4, PO.19.00401. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Kurzawinski, T.; Ross, E.; Stoneham, S.; Beale, T.; Proctor, I.; Hulse, T.; Simpson, K.; Gaze, M.N.; Cattaneo, E.; et al. Treatment Outcome with a Selective RET Tyrosine Kinase Inhibitor Selpercatinib in Children with Multiple Endocrine Neoplasia Type 2 and Advanced Medullary Thyroid Carcinoma. Eur. J. Cancer Oxf. Engl. 2021, 158, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gainor, J.F.; Oxnard, G.R.; Tan, D.S.W.; Owen, D.H.; Cho, B.C.; Loong, H.H.; McCoach, C.E.; Weiss, J.; Kim, Y.J.; et al. Intracranial Efficacy of Selpercatinib in RET Fusion-Positive Non-Small Cell Lung Cancers on the LIBRETTO-001 Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Andreev-Drakhlin, A.; Cabanillas, M.; Amini, B.; Subbiah, V. Systemic and CNS Activity of Selective RET Inhibition With Selpercatinib (LOXO-292) in a Patient With RET-Mutant Medullary Thyroid Cancer With Extensive CNS Metastases. JCO Precis. Oncol. 2020, 4, PO.20.00096. [Google Scholar] [CrossRef]
- Tsui, D.C.C.; Kavanagh, B.D.; Honce, J.M.; Rossi, C.; Patil, T.; Camidge, D.R. Central Nervous System Response to Selpercartinib in Patient With RET-Rearranged Non-Small Cell Lung Cancer After Developing Leptomeningeal Disease on Pralsetinib. Clin. Lung Cancer 2022, 23, e5–e8. [Google Scholar] [CrossRef]
- Drilon, A.E.; Zhai, D.; Rogers, E.; Deng, W.; Zhang, X.; Ung, J.; Lee, D.; Rodon, L.; Graber, A.; Zimmerman, Z.F.; et al. The Next-Generation RET Inhibitor TPX-0046 Is Active in Drug-Resistant and Naïve RET-Driven Cancer Models. J. Clin. Oncol. 2020, 38, 3616. [Google Scholar] [CrossRef]
- Schoffski, P.; Cho, B.C.; Italiano, A.; Loong, H.H.F.; Massard, C.; Medina Rodriguez, L.; Shih, J.-Y.; Subbiah, V.; Verlingue, L.; Andreas, K.; et al. BOS172738, a Highly Potent and Selective RET Inhibitor, for the Treatment of RET-Altered Tumors Including RET-Fusion+ NSCLC and RET-Mutant MTC: Phase 1 Study Results. J. Clin. Oncol. 2021, 39, 3008. [Google Scholar] [CrossRef]
- Miyazaki, I.; Ishida, K.; Kato, M.; Suzuki, T.; Fujita, H.; Ohkubo, S.; Iwasawa, Y. Abstract P06-02: Discovery of TAS0953/HM06, a Novel next Generation RET-Specific Inhibitor Capable of Inhibiting RET Solvent Front Mutations. Mol. Cancer Ther. 2021, 20, P06-02. [Google Scholar] [CrossRef]
- Kolakowski, G.R.; Anderson, E.D.; Ballard, J.A.; Brandhuber, B.J.; Condroski, K.R.; Gomez, E.B.; Irvin, T.C.; Kumar, M.; Patel, N.A.; Watson, F.D.; et al. Abstract 1464: Pre-Clinical Characterization of Potent and Selective next-Generation RET Inhibitors. Cancer Res. 2021, 81, 1464. [Google Scholar] [CrossRef]
- Liu, X.; Shen, T.; Mooers, B.H.M.; Hilberg, F.; Wu, J. Drug Resistance Profiles of Mutations in the RET Kinase Domain. Br. J. Pharmacol. 2018, 175, 3504–3515. [Google Scholar] [CrossRef]
- Lu, C.; Zhou, Q. Diagnostics, Therapeutics and RET Inhibitor Resistance for RET Fusion-Positive Non-Small Cell Lung Cancers and Future Perspectives. Cancer Treat. Rev. 2021, 96, 102153. [Google Scholar] [CrossRef]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.; Curigliano, G.; Doebele, R.C.; Lin, J.J.; Ou, S.-H.; Miller, S.; Turner, C.D.; Subbiah, V. OA05.02 Analysis of Resistance Mmechanisms to Pralsetinib in Patients with RET Fusion-Positive Non-Small Cell Lung Cancer (NSCLC) from the ARROW Study. J. Thorac. Oncol. 2021, 16, S5. [Google Scholar] [CrossRef]
- Drilon, A.; Rogers, E.; Zhai, D.; Deng, W.; Zhang, X.; Lee, D.; Ung, J.; Whitten, J.; Zhang, H.; Liu, J.; et al. TPX-0046 Is a Novel and Potent RET/SRC Inhibitor for RET-Driven Cancers. Ann. Oncol. 2019, 30, v190–v191. [Google Scholar] [CrossRef]
- Repetto, M.; Crimini, E.; Ascione, L.; Boscolo Bielo, L.; Belli, C.; Curigliano, G. The Return of RET GateKeeper Mutations? An in-Silico Exploratory Analysis of Potential Resistance Mechanisms to Novel RET Macrocyclic Inhibitor TPX-0046. Investig. New Drugs 2022, 40, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Hu, X.; Liu, X.; Subbiah, V.; Mooers, B.H.M.; Wu, J. The L730V/I RET Roof Mutations Display Different Activities toward Pralsetinib and Selpercatinib. NPJ Precis. Oncol. 2021, 5, 48. [Google Scholar] [CrossRef]
- Subbiah, V.; Shen, T.; Tetzlaff, M.; Weissferdt, A.; Byers, L.A.; Cascone, T.; Behrang, A.; Meric-Bernstam, F.; Mooers, B.H.M.; Rothenberg, S.M.; et al. Patient-Driven Discovery and Post-Clinical Validation of NTRK3 Fusion as an Acquired Resistance Mechanism to Selpercatinib in RET Fusion-Positive Lung Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 817–819. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Won, H.H.; Zheng, Y.; Cocco, E.; Selcuklu, D.; Gong, Y.; Friedman, N.D.; de Bruijn, I.; Sumer, O.; Bielski, C.M.; et al. The Evolution of RET Inhibitor Resistance in RET-Driven Lung and Thyroid Cancers. Nat. Commun. 2022, 13, 1450. [Google Scholar] [CrossRef]
- Lin, J.J.; Liu, S.V.; McCoach, C.E.; Zhu, V.W.; Tan, A.C.; Yoda, S.; Peterson, J.; Do, A.; Prutisto-Chang, K.; Dagogo-Jack, I.; et al. Mechanisms of Resistance to Selective RET Tyrosine Kinase Inhibitors in RET Fusion-Positive Non-Small-Cell Lung Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1725–1733. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Johnson, M.L.; Clifford, S.E.; Somwar, R.; Kherani, J.F.; Son, J.; Bertram, A.A.; Davare, M.A.; Gladstone, E.; Ivanova, E.V.; et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion-Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 34–42. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Selpercatinib (LOXO-292) | Pralsetinib (BLU-667) | |
|---|---|---|---|
| Trial | LIBRETTO-001/Multicenter, Phase 1/2 | ARROW/Multicenter, Phase 1/2 | |
| Date of FDA approval/type of approval | 8 May 2020 for Adult patients with metastatic, RET fusion-positive NSCLC Adult and pediatric (≥12 years of age) patients with
| 4 September 2020 for Adult patients with metastatic RET fusion-positive NSCLC 1 December 2020 for Adult and pediatric (≥12 years of age) patients with
| |
| Clinical trial results | NSCLC | RET-positive NSCLC patients [90]
| RET-positive NSCLC patients [82]
|
| Thyroid cancer | RET-positive thyroid cancer patients [85]
| RET-positive thyroid cancer patients [84]
| |
| Adverse events | NSCLC arm Grade 1 & 2—39%, grade 3 & 4—58% Thyroid cancer arm Grade 1& 2—32%, grade 3 & 4—66% | NSCLC arm All toxicities—93%, grade 3 & 4—48% Thyroid cancer arm Grade 1 & 2—43%, grade 3 & 4—53% | |
| Most common grade 3 & 4 AEs related to treatment | NSCLC | Hypertension (9%), increased ALT (9%), increased AST (6%), diarrhea (2%), prolonged QT (2%), constipation (1%), rash (1%), vomiting (1%), pyrexia (1%), thrombocytopenia (1%) | Neutropenia (19%), hypertension (11%), anemia (10%), decreased white blood cell count (6%), lymphopenia (5%), increased ALT (3%), increased AST (3%), phosphokinase (3%). thrombocytopenia (3%), Elevated blood creatine phosphokinase (3%), pneumonia (<4%) |
| Thyroid cancer | Hypertension (12%), increased ALT (11%), increased AST (8%), diarrhea (3%), prolonged QT (2%), fatigue (1%), headache (1%), weight increased (1%) | Hypertension (17%), neutropenia (14%), lymphopenia (12%), anemia (10%), decreased white blood cell count (8%), increased blood creatine phosphokinase (5%), asthenia (4%), pneumonitis (3%), diarrhoea (2%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucharczyk, T.; Krawczyk, P.; Kowalski, D.M.; Płużański, A.; Kubiatowski, T.; Kalinka, E. RET Proto-Oncogene—Not Such an Obvious Starting Point in Cancer Therapy. Cancers 2022, 14, 5298. https://doi.org/10.3390/cancers14215298
Kucharczyk T, Krawczyk P, Kowalski DM, Płużański A, Kubiatowski T, Kalinka E. RET Proto-Oncogene—Not Such an Obvious Starting Point in Cancer Therapy. Cancers. 2022; 14(21):5298. https://doi.org/10.3390/cancers14215298
Chicago/Turabian StyleKucharczyk, Tomasz, Paweł Krawczyk, Dariusz M. Kowalski, Adam Płużański, Tomasz Kubiatowski, and Ewa Kalinka. 2022. "RET Proto-Oncogene—Not Such an Obvious Starting Point in Cancer Therapy" Cancers 14, no. 21: 5298. https://doi.org/10.3390/cancers14215298