
Factors That Affect the Formation of Chromosomal Translocations in Cells
 , , , , and
, , , , and 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. History of Chromosomal Translocations in Cancer
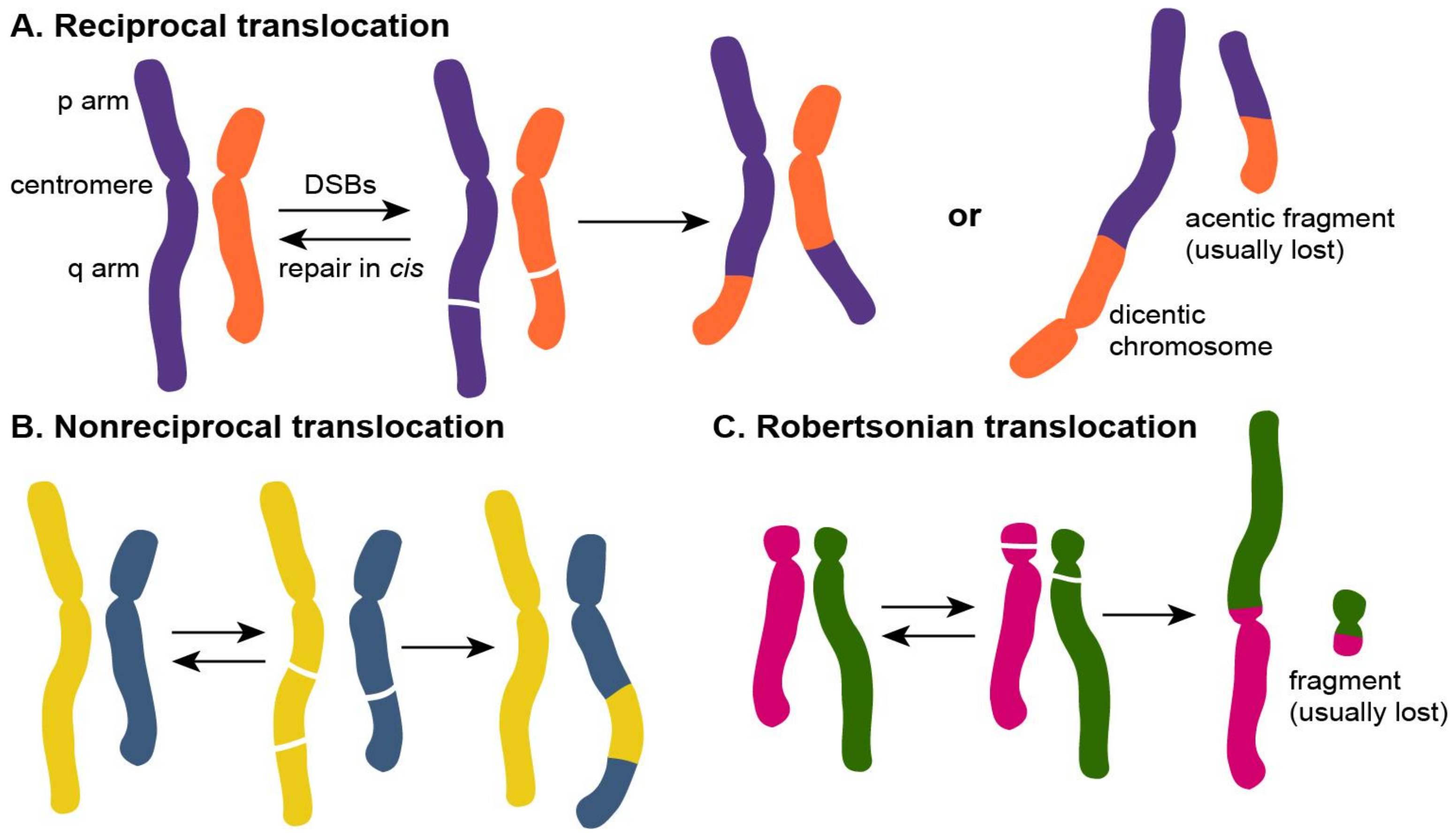
1.2. Types of Chromosomal Translocations
1.3. Chromosomal Translocations Affect the Normal Cell Functions
2. Sources of DSBs In Vivo and Their Experimental Modeling for Studying Translocations
2.1. Sources of DSBs
2.2. Experimental Models of Chromosomal Translocations
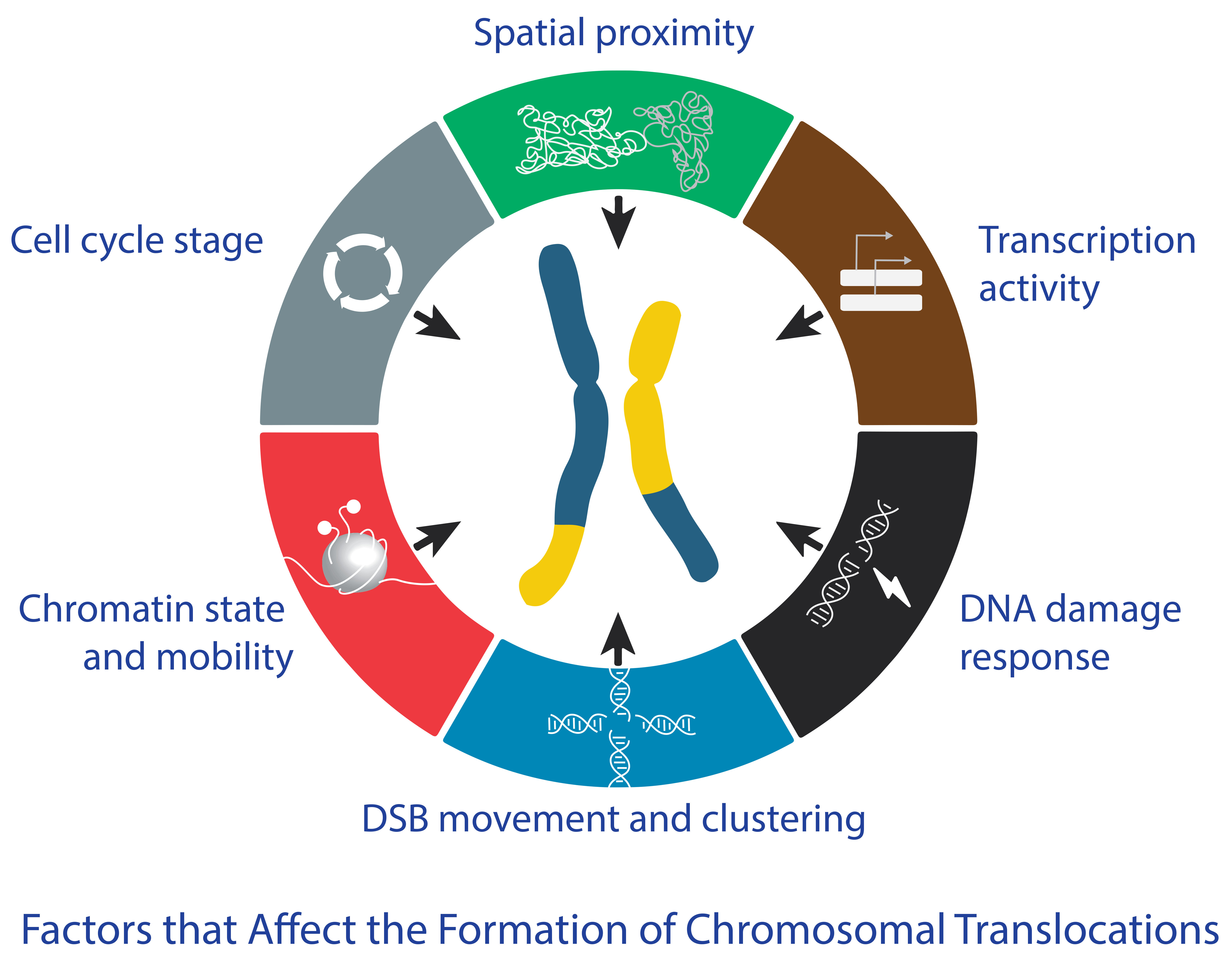
3. How Do the Different Factors Affect the Formation of Chromosomal Translocations?
3.1. Spatial Proximity
3.2. Transcriptional Activity
3.3. DNA Damage Response
3.4. Chromatin State and Mobility
3.5. DSB Movement and Clustering
3.6. Cell Cycle
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nussenzweig, A.; Nussenzweig, M.C. Origin of Chromosomal Translocations in Lymphoid Cancer. Cell 2010, 141, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, D.A.; Nussenzweig, A. Mechanisms Driving Chromosomal Translocations: Lost in Time and Space. Oncogene 2021, 40, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Roukos, V.; Misteli, T. The Biogenesis of Chromosome Translocations. Nat. Cell Biol. 2014, 16, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Oncogenic Chromosomal Translocations and Human Cancer (Review). Oncol. Rep. 2013, 30, 2011–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmain, A. Cancer Genetics: From Boveri and Mendel to Microarrays. Nat. Rev. Cancer 2001, 1, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and Annotated by Henry Harris. J. Cell Sci. 2008, 121, 1–84. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [CrossRef]
- Levan, A. Some Current Problems of Cancer Cytogenetics. Hereditas 1967, 57, 343–355. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia Identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Rowley, J.D. Identification of a Translocation with Quinacrine Fluorescence in a Patient with Acute Leukemia. Ann. De Genet. 1973, 16, 109–112. [Google Scholar]
- Bohlander, S.K.; Kakadiya, P.M.; Coysh, A. Chromosome Rearrangements and Translocations. In Encyclopedia of Cancer, 3rd ed.; Boffetta, P., Hainaut, P., Eds.; Academic Press: Oxford, UK, 2019; pp. 389–404. ISBN 978-0-12-812485-7. [Google Scholar]
- Morin, S.J.; Eccles, J.; Iturriaga, A.; Zimmerman, R.S. Translocations, Inversions and Other Chromosome Rearrangements. Fertil. Steril. 2017, 107, 19–26. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The Dark Side of Centromeres: Types, Causes and Consequences of Structural Abnormalities Implicating Centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [Green Version]
- Robertson, W.R.B. Chromosome Studies. I. Taxonomic Relationships Shown in the Chromosomes of Tettigidae and Acrididae: V-Shaped Chromosomes and Their Significance in Acrididae, Locustidae, and Gryllidae: Chromosomes and Variation. J. Morphol. 1916, 27, 179–331. [Google Scholar] [CrossRef] [Green Version]
- Matveevsky, S.; Tretiakov, A.; Kashintsova, A.; Bakloushinskaya, I.; Kolomiets, O. Meiotic Nuclear Architecture in Distinct Mole Vole Hybrids with Robertsonian Translocations: Chromosome Chains, Stretched Centromeres, and Distorted Recombination. Int. J. Mol. Sci. 2020, 21, 7630. [Google Scholar] [CrossRef]
- Spinner, N.B.; Conlin, L.K.; Mulchandani, S.; Emanuel, B.S. Chapter 45—Deletions and Other Structural Abnormalities of the Autosomes. In Emery and Rimoin’s Principles and Practice of Medical Genetics, 6th ed.; Rimoin, D., Pyeritz, R., Korf, B., Eds.; Academic Press: Oxford, UK, 2013; pp. 1–37. ISBN 978-0-12-383834-6. [Google Scholar]
- Eykelenboom, J.E.; Briggs, G.J.; Bradshaw, N.J.; Soares, D.C.; Ogawa, F.; Christie, S.; Malavasi, E.L.V.; Makedonopoulou, P.; Mackie, S.; Malloy, M.P.; et al. A t(1;11) Translocation Linked to Schizophrenia and Affective Disorders Gives Rise to Aberrant Chimeric DISC1 Transcripts That Encode Structurally Altered, Deleterious Mitochondrial Proteins. Hum. Mol. Genet. 2012, 21, 3374–3386. [Google Scholar] [CrossRef] [Green Version]
- Fraccaro, M.; Maraschio, P.; Pasquali, F.; Tiepolo, L.; Zuffardi, O.; Giarola, A. Male Infertility and 13–14 Translocation. Lancet 1973, 1, 488. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Available online: https://mitelmandatabase.isb-cgc.org (accessed on 5 July 2021).
- Nakano, K.; Takahashi, S. Translocation-Related Sarcomas. Int. J. Mol. Sci. 2018, 19, 3784. [Google Scholar] [CrossRef] [Green Version]
- Prasher, V.P. Presenile Dementia Associated with Unbalanced Robertsonian Translocation Form of Down’s Syndrome. Lancet 1993, 342, 686–687. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Wang, R.-X.; Pan, Y.; Zhang, H.; Li, L.-L.; Zhu, H.-B.; Liu, R.-Z. A Report of Nine Cases and Review of the Literature of Infertile Men Carrying Balanced Translocations Involving Chromosome 5. Mol. Cytogenet 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Daley, G.Q.; Baltimore, D. Transformation of an Interleukin 3-Dependent Hematopoietic Cell Line by the Chronic Myelogenous Leukemia-Specific P210(Bcr/Abl) Protein. Proc. Natl. Acad. Sci. USA 1988, 85, 9312–9316. [Google Scholar] [CrossRef] [Green Version]
- Daley, G.Q.; Van Etten, R.A.; Baltimore, D. Induction of Chronic Myelogenous Leukemia in Mice by the P210 Bcr/Abl Gene of the Philadelphia Chromosome. Science 1990, 247, 824–830. [Google Scholar] [CrossRef]
- Gishizky, M.L.; Johnson-White, J.; Witte, O.N. Efficient Transplantation of BCR-ABL-Induced Chronic Myelogenous Leukemia- like Syndrome in Mice. Proc. Natl. Acad. Sci. USA 1993, 90, 3755–3759. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The C-Myc Oncogene Driven by Immunoglobulin Enhancers Induces Lymphoid Malignancy in Transgenic Mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef]
- Ar-Rushdi, A.; Nishikura, K.; Erikson, J.; Watt, R.; Rovera, G.; Croce, C.M. Differential Expression of the Translocated and the Untranslocated C-Myc Oncogene in Burkitt Lymphoma. Science 1983, 222, 390–393. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human C-Myc Onc Gene Is Located on the Region of Chromosome 8 That Is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [Green Version]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the C-Myc Gene into the Immunoglobulin Heavy Chain Locus in Human Burkitt Lymphoma and Murine Plasmacytoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [Green Version]
- Zech, L.; Haglund, U.; Nilsson, K.; Klein, G. Characteristic Chromosomal Abnormalities in Biopsies and Lymphoid-cell Lines from Patients with Burkitt and Non-burkitt Lymphomas. Int. J. Cancer 1976, 17, 47–56. [Google Scholar] [CrossRef]
- Allinne, J.; Pichugin, A.; Iarovaia, O.; Klibi, M.; Barat, A.; Zlotek-Zlotkiewicz, E.; Markozashvili, D.; Petrova, N.; Camara-Clayette, V.; Ioudinkova, E.; et al. Perinucleolar Relocalization and Nucleolin as Crucial Events in the Transcriptional Activation of Key Genes in Mantle Cell Lymphoma. Blood 2014, 123, 2044–2053. [Google Scholar] [CrossRef] [Green Version]
- Bickmore, W.A.; van Steensel, B. Genome Architecture: Domain Organization of Interphase Chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Harewood, L.; Schutz, F.; Boyle, S.; Perry, P.; Delorenzi, M.; Bickmore, W.A.; Reymond, A. The Effect of Translocation-Induced Nuclear Reorganization on Gene Expression. Genome Res. 2010, 20, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Meaburn, K.J. Spatial Genome Organization and Its Emerging Role as a Potential Diagnosis Tool. Front. Genet. 2016, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Lieber, M.R. Concept of DNA Lesion Longevity and Chromosomal Translocations. Trends Biochem. Sci. 2018, 43, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xie, W. The Role of 3D Genome Organization in Development and Cell Differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Nussenzweig, M.C. Chromosome Translocation, B Cell Lymphoma, and Activation-Induced Cytidine Deaminase. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 79–103. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Lobato, M.N.; Metzler, M.; Drynan, L.; Forster, A.; Pannell, R.; Rabbitts, T.H. Modeling Chromosomal Translocations Using Conditional Alleles to Recapitulate Initiating Events in Human Leukemias. JNCI Monogr. 2008, 2008, 58–63. [Google Scholar] [CrossRef]
- Nambiar, M.; Raghavan, S.C. How Does DNA Break during Chromosomal Translocations? Nucleic Acids Res. 2011, 39, 5813–5825. [Google Scholar] [CrossRef] [Green Version]
- Mitelman, F.; Johansson, B.; Mertens, F. Mitelman Database Chromosome Aberrations and Gene Fusions in Cancer. Available online: https://mitelmandatabase.isb-cgc.org/about (accessed on 28 January 2020).
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G. Processing of DNA Damage Clusters in Human Cells: Current Status of Knowledge. Mol. BioSystems 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Sax, K. Chromosome Aberrations Induced by X-Rays. Genetics 1938, 23, 494–516. [Google Scholar] [CrossRef]
- Shikazono, N.; Noguchi, M.; Fujii, K.; Urushibara, A.; Yokoya, A. The Yield, Processing, and Biological Consequences of Clustered DNA Damage Induced by Ionizing Radiation. J. Radiat Res. 2009, 50, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.M.; Bennett, P.V.; Sutherland, J.C.; Laval, J. Clustered DNA Damages Induced by X Rays in Human Cells. Rare 2002, 157, 611–616. [Google Scholar] [CrossRef]
- Ward, J.F. DNA Damage Produced by Ionizing Radiation in Mammalian Cells: Identities, Mechanisms of Formation, and Reparability. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Moldave, K., Eds.; Academic Press: Cambridge, MA, USA, 1988; Volume 35, pp. 95–125. [Google Scholar]
- Cao, L.; Alani, E.; Kleckner, N. A Pathway for Generation and Processing of Double-Strand Breaks during Meiotic Recombination in S. Cerevisiae. Cell 1990, 61, 1089–1101. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-Specific DNA Double-Strand Breaks Are Catalyzed by Spo11, a Member of a Widely Conserved Protein Family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [Green Version]
- De Massy, B. Initiation of Meiotic Recombination: How and Where? Conservation and Specificities Among Eukaryotes. Annu. Rev. Genet. 2013, 47, 563–599. [Google Scholar] [CrossRef]
- Paigen, K.; Petkov, P. Mammalian Recombination Hot Spots: Properties, Control and Evolution. Nat. Rev. Genet. 2010, 11, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Cobb, R.M.; Oestreich, K.J.; Osipovich, O.A.; Oltz, E.M. Accessibility Control of V(D)J Recombination. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2006; Volume 91, pp. 45–109. [Google Scholar]
- Davis, M.M.; Bjorkman, P.J. T-Cell Antigen Receptor Genes and T-Cell Recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Krangel, M.S. Mechanics of T Cell Receptor Gene Rearrangement. Curr. Opin. Immunol. 2009, 21, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, J.; Basu, U.; Zarrin, A.; Yan, C.; Franco, S.; Perlot, T.; Vuong, B.; Wang, J.; Phan, R.T.; Datta, A.; et al. Evolution of the Immunoglobulin Heavy Chain Class Switch Recombination Mechanism. In Advances in Immunology; AID for Immunoglobulin Diversity; Academic Press: Cambridge, MA, USA, 2007; Volume 94, pp. 157–214. [Google Scholar]
- Di Noia, J.M.; Neuberger, M.S. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef]
- Schatz, D.G.; Baltimore, D. Uncovering the V(D)J Recombinase. Cell 2004, 116, S103–S108. [Google Scholar] [CrossRef] [Green Version]
- Honjo, T.; Kinoshita, K.; Muramatsu, M. Molecular Mechanism of Class Switch Recombination: Linkage with Somatic Hypermutation. Annu. Rev. Immunol. 2002, 20, 165–196. [Google Scholar] [CrossRef]
- Liu, M.; Duke, J.L.; Richter, D.J.; Vinuesa, C.G.; Goodnow, C.C.; Kleinstein, S.H.; Schatz, D.G. Two Levels of Protection for the B Cell Genome during Somatic Hypermutation. Nature 2008, 451, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common Fragile Sites Are Conserved Features of Human and Mouse Chromosomes and Relate to Large Active Genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, P.; Aguilera, A. Cotranscriptionally Formed DNA:RNA Hybrids Mediate Transcription Elongation Impairment and Transcription-Associated Recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef]
- Iannelli, F.; Galbiati, A.; Capozzo, I.; Nguyen, Q.; Magnuson, B.; Michelini, F.; D’Alessandro, G.; Cabrini, M.; Roncador, M.; Francia, S.; et al. A Damaged Genome’s Transcriptional Landscape through Multilayered Expression Profiling around in Situ -Mapped DNA Double-Strand Breaks. Nat. Commun. 2017, 8, 15656. [Google Scholar] [CrossRef] [Green Version]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.-M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular Profiling of Common Fragile Sites in Human Fibroblasts. Nat. Struct. Mol. Biol. 2011, 18, 1421–1423. [Google Scholar] [CrossRef]
- Sutherland, G.R. Heritable Fragile Sites on Human Chromosomes II. Distribution, Phenotypic Effects, and Cytogenetics. Am. J. Hum. Genet. 1979, 31, 136–148. [Google Scholar]
- Pfeiffer, P.; Goedecke, W.; Obe, G. Mechanisms of DNA Double-Strand Break Repair and Their Potential to Induce Chromosomal Aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef]
- Syeda, A.H.; Hawkins, M.; McGlynn, P. Recombination and Replication. Cold Spring Harb Perspect Biol. 2014, 6, a016550. [Google Scholar] [CrossRef]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative Damage to DNA during Aging: 8-Hydroxy-2’-Deoxyguanosine in Rat Organ DNA and Urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T.; Nyberg, B. Rate of Depurination of Native Deoxyribonucleic Acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef]
- Jain, S.; Shukla, S.; Yang, C.; Zhang, M.; Fatma, Z.; Lingamaneni, M.; Abesteh, S.; Lane, S.T.; Xiong, X.; Wang, Y.; et al. TALEN Outperforms Cas9 in Editing Heterochromatin Target Sites. Nat. Commun. 2021, 12, 606. [Google Scholar] [CrossRef]
- Mitrentsi, I.; Soutoglou, E. CRISPR/Cas9-Induced Breaks in Heterochromatin, Visualized by Immunofluorescence. Methods Mol. Biol. 2021, 2153, 439–445. [Google Scholar] [CrossRef]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef]
- Iarovaia, O.V.; Rubtsov, M.; Ioudinkova, E.; Tsfasman, T.; Razin, S.V.; Vassetzky, Y.S. Dynamics of Double Strand Breaks and Chromosomal Translocations. Mol. Cancer 2014, 13, 249. [Google Scholar] [CrossRef] [Green Version]
- Brunet, E.; Simsek, D.; Tomishima, M.; DeKelver, R.; Choi, V.M.; Gregory, P.; Urnov, F.; Weinstock, D.M.; Jasin, M. Chromosomal Translocations Induced at Specified Loci in Human Stem Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10620–10625. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Saada, Y.B.; Tsfasman, T.; Osina, K.; Robin, C.; Lomov, N.; Rubtsov, M.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A One-Step PCR-Based Assay to Evaluate the Efficiency and Precision of Genomic DNA-Editing Tools. Mol. Ther. Methods Clin. Dev. 2017, 5, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Piganeau, M.; Ghezraoui, H.; De Cian, A.; Guittat, L.; Tomishima, M.; Perrouault, L.; René, O.; Katibah, G.E.; Zhang, L.; Holmes, M.C.; et al. Cancer Translocations in Human Cells Induced by Zinc Finger and TALE Nucleases. Genome Res. 2013, 23, 1182–1193. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Martin, M.C.; Garcia, A.; Cigudosa, J.C.; Ramirez, J.C.; Rodriguez-Perales, S. Engineering Human Tumour-Associated Chromosomal Translocations with the RNA-Guided CRISPR–Cas9 System. Nat. Commun. 2014, 5, 3964. [Google Scholar] [CrossRef]
- Vanoli, F.; Jasin, M. Generation of Chromosomal Translocations That Lead to Conditional Fusion Protein Expression Using CRISPR-Cas9 and Homology-Directed Repair. Methods 2017, 121–122, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Shmakova, A.A.; Germini, D.; Vassetzky, Y.S. Exploring the Features of Burkitt’s Lymphoma-Associated t(8;14) Translocations Generated via a CRISPR/Cas9-Based System. Biopolym. Cell 2019, 35, 232–233. [Google Scholar] [CrossRef]
- Canoy, R.J.; André, F.; Shmakova, A.; Wiels, J.; Lipinski, M.; Vassetzky, Y.; Germini, D. Easy and Robust Electrotransfection Protocol for Efficient Ectopic Gene Expression and Genome Editing in Human B Cells. Gene Ther. 2020, 1–5. [Google Scholar] [CrossRef]
- Shmakova, A.A.; Lomov, N.; Viushkov, V.; Tsfasman, T.; Kozhevnikova, Y.; Sokolova, D.; Pokrovsky, V.; Syrkina, M.; Germini, D.; Rubtsov, M.; et al. Cell Models with Inducible Oncogenic Translocations Allow to Evaluate the Potential of Drugs to Favor Secondary Translocations. Cancer Commun. 2022, in press. [Google Scholar] [CrossRef]
- Shmakova, A.A.; Shmakova, O.P.; Karpukhina, A.A.; Vassetzky, Y.S. CRISPR/Cas: History and Perspectives. Russ. J. Dev. Biol. 2022, 53, 272–282. [Google Scholar] [CrossRef]
- Byrne, M.; Wray, J.; Reinert, B.; Wu, Y.; Nickoloff, J.; Lee, S.-H.; Hromas, R.; Williamson, E. Mechanisms of Oncogenic Chromosomal Translocations. Ann. New York Acad. Sci. 2014, 1310, 89–97. [Google Scholar] [CrossRef]
- Lieber, M.R. Mechanisms of Human Lymphoid Chromosomal Translocations. Nat. Rev. Cancer 2016, 16, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Zhang, Y.; Frock, R.L.; Lewis, S.M.; Molinie, B.; Ho, Y.J.; Myers, D.R.; Choi, V.W.; Compagno, M.; Malkin, D.J.; et al. Genome-Wide Translocation Sequencing Reveals Mechanisms of Chromosome Breaks and Rearrangements in B Cells. Cell 2011, 147, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Klein, I.A.; Resch, W.; Jankovic, M.; Oliveira, T.; Yamane, A.; Nakahashi, H.; Di Virgilio, M.; Bothmer, A.; Nussenzweig, A.; Robbiani, D.F.; et al. Translocation-Capture Sequencing Reveals the Extent and Nature of Chromosomal Rearrangements in B Lymphocytes. Cell 2011, 147, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-Induced TOP2B-Mediated Double-Strand Breaks and Prostate Cancer Gene Rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.-M.; et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 2019, 75, 252–266.e8. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [Green Version]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M.; Dietzel, S.; Müller, S.; Solovei, I.; Fakan, S. Chromosome Territories—A Functional Nuclear Landscape. Curr. Opin. Cell Biol. 2006, 18, 307–316. [Google Scholar] [CrossRef]
- Boveri, T. Die Blastomerenkerne von Ascaris Megalocephala Und Die Theorie Der Chromosomenindividualität. Arch. Zellforsch 1909, 3, 181–268. [Google Scholar]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb Perspect Biol. 2010, 2, a003889. [Google Scholar] [CrossRef] [Green Version]
- Belmont, A.S.; Bruce, K. Visualization of G1 Chromosomes: A Folded, Twisted, Supercoiled Chromonema Model of Interphase Chromatid Structure. J. Cell Biol. 1994, 127, 287–302. [Google Scholar] [CrossRef]
- Marella, N.V.; Bhattacharya, S.; Mukherjee, L.; Xu, J.; Berezney, R. Cell Type Specific Chromosome Territory Organization in the Interphase Nucleus of Normal and Cancer Cells. J. Cell. Physiol. 2009, 221, 130–138. [Google Scholar] [CrossRef]
- Parada, L.A.; McQueen, P.G.; Misteli, T. Tissue-Specific Spatial Organization of Genomes. Genome Biol. 2004, 5, R44. [Google Scholar] [CrossRef] [Green Version]
- Roix, J.J.; McQueen, P.G.; Munson, P.J.; Parada, L.A.; Misteli, T. Spatial Proximity of Translocation-Prone Gene Loci in Human Lymphomas. Nat. Genet. 2003, 34, 287–291. [Google Scholar] [CrossRef]
- Haber, J.E.; Leung, W.-Y. Lack of Chromosome Territoriality in Yeast: Promiscuous Rejoining of Broken Chromosome Ends. Proc. Natl. Acad. Sci. USA 1996, 93, 13949–13954. [Google Scholar] [CrossRef]
- Fraser, J.; Williamson, I.; Bickmore, W.A.; Dostie, J. An Overview of Genome Organization and How We Got There: From FISH to Hi-C. Microbiol. Mol. Biol. Rev. 2015, 79, 347–372. [Google Scholar] [CrossRef] [Green Version]
- Fritz, A.J.; Barutcu, A.R.; Martin-Buley, L.; van Wijnen, A.J.; Zaidi, S.K.; Imbalzano, A.N.; Lian, J.B.; Stein, J.L.; Stein, G.S. Chromosomes at Work: Organization of Chromosome Territories in the Interphase Nucleus. J. Cell. Biochem. 2016, 117, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Lukášová, E.; Kozubek, S.; Kozubek, M.; Kjeronská, J.; Rýznar, L.; Horáková, J.; Krahulcová, E.; Horneck, G. Localisation and Distance between ABL and BCR Genes in Interphase Nuclei of Bone Marrow Cells of Control Donors and Patients with Chronic Myeloid Leukaemia. Hum. Genet. 1997, 100, 525–535. [Google Scholar] [CrossRef]
- Neves, H.; Ramos, C.; da Silva, M.G.; Parreira, A.; Parreira, L. The Nuclear Topography of ABL, BCR, PML, and RARalpha Genes: Evidence for Gene Proximity in Specific Phases of the Cell Cycle and Stages of Hematopoietic Differentiation. Blood 1999, 93, 1197–1207. [Google Scholar] [CrossRef]
- Boxer, L.M.; Dang, C.V. Translocations Involving C- Myc and c- Myc Function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Gostissa, M.; Ranganath, S.; Bianco, J.M.; Alt, F.W. Chromosomal Location Targets Different MYC Family Gene Members for Oncogenic Translocations. Proc. Natl. Acad. Sci. USA 2009, 106, 2265–2270. [Google Scholar] [CrossRef] [Green Version]
- Sathitruangsak, C.; Righolt, C.H.; Klewes, L.; Chang, D.T.; Kotb, R.; Mai, S. Distinct and Shared Three-Dimensional Chromosome Organization Patterns in Lymphocytes, Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma. Int. J. Cancer 2017, 140, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; McCord, R.P.; Ho, Y.-J.; Lajoie, B.R.; Hildebrand, D.G.; Simon, A.C.; Becker, M.S.; Alt, F.W.; Dekker, J. Spatial Organization of the Mouse Genome and Its Role in Recurrent Chromosomal Translocations. Cell 2012, 148, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-Resolution DNA Double-Strand Breaks Mapping by next-Generation Sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Lensing, S.V.; Marsico, G.; Hänsel-Hertsch, R.; Lam, E.Y.; Tannahill, D.; Balasubramanian, S. DSBCapture: In Situ Capture and Sequencing of DNA Breaks. Nat. Methods 2016, 13, 855–857. [Google Scholar] [CrossRef]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin Structure Influences the Sensitivity of DNA to Gamma-Radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A. The Connection between Transcription and Genomic Instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R Loops: New Modulators of Genome Dynamics and Function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Stirling, P.C.; Hieter, P. Canonical DNA Repair Pathways Influence R-Loop-Driven Genome Instability. J. Mol. Biol. 2017, 429, 3132–3138. [Google Scholar] [CrossRef] [PubMed]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations Involving 8q24 in Burkitt Lymphoma and Other Malignant Lymphomas: A Historical Review of Cytogenetics in the Light of Todays Knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquette, M.L.; Pham, P.; Goodman, M.F.; Maizels, N. AID Binds to Transcription-Induced Structures in c- MYC That Map to Regions Associated with Translocation and Hypermutation. Oncogene 2005, 24, 5791–5798. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Chedin, F.; Hsieh, C.-L.; Wilson, T.E.; Lieber, M.R. R-Loops at Immunoglobulin Class Switch Regions in the Chromosomes of Stimulated B Cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef]
- Pavri, R.; Gazumyan, A.; Jankovic, M.; Di Virgilio, M.; Klein, I.; Ansarah-Sobrinho, C.; Resch, W.; Yamane, A.; Reina San-Martin, B.; Barreto, V.; et al. Activation-Induced Cytidine Deaminase Targets DNA at Sites of RNA Polymerase II Stalling by Interaction with Spt5. Cell 2010, 143, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Yamane, A.; Resch, W.; Kuo, N.; Kuchen, S.; Li, Z.; Sun, H.; Robbiani, D.F.; McBride, K.; Nussenzweig, M.C.; Casellas, R. Deep-Sequencing Identification of the Genomic Targets of the Cytidine Deaminase AID and Its Cofactor RPA in B Lymphocytes. Nat. Immunol. 2011, 12, 62–69. [Google Scholar] [CrossRef]
- Yang, Y.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Ramiro, A.R.; Jankovic, M.; Callen, E.; Difilippantonio, S.; Chen, H.-T.; McBride, K.M.; Eisenreich, T.R.; Chen, J.; Dickins, R.A.; Lowe, S.W.; et al. Role of Genomic Instability and P53 in AID-Induced c-Myc—Igh Translocations. Nature 2006, 440, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Yeo, A.J.; Becherel, O.J.; Luff, J.E.; Cullen, J.K.; Wongsurawat, T.; Jenjaroenpoon, P.; Kuznetsov, V.A.; McKinnon, P.J.; Lavin, M.F. R-Loops in Proliferating Cells but Not in the Brain: Implications for AOA2 and Other Autosomal Recessive Ataxias. PLoS ONE 2014, 9, e90219. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 Prevents R-Loop Accumulation and Associates with TREX-2 MRNA Export Factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in Human Cancer Genomes: Footprints and Mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Pliss, A.; Malyavantham, K.; Bhattacharya, S.; Zeitz, M.; Berezney, R. Chromatin Dynamics Is Correlated with Replication Timing. Chromosoma 2009, 118, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.S.; Chakalova, L.; Brown, K.E.; Carter, D.; Horton, A.; Debrand, E.; Goyenechea, B.; Mitchell, J.A.; Lopes, S.; Reik, W.; et al. Active Genes Dynamically Colocalize to Shared Sites of Ongoing Transcription. Nat. Genet. 2004, 36, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.S.; Chakalova, L.; Mitchell, J.A.; Horton, A.; Wood, A.L.; Bolland, D.J.; Corcoran, A.E.; Fraser, P. Myc Dynamically and Preferentially Relocates to a Transcription Factory Occupied by Igh. PLoS Biol. 2007, 5, e192. [Google Scholar] [CrossRef]
- Barlow, J.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. A Novel Class of Early Replicating Fragile Sites That Contribute to Genome Instability in B Cell Lymphomas. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef]
- Mathas, S.; Kreher, S.; Meaburn, K.J.; Jöhrens, K.; Lamprecht, B.; Assaf, C.; Sterry, W.; Kadin, M.E.; Daibata, M.; Joos, S.; et al. Gene Deregulation and Spatial Genome Reorganization near Breakpoints Prior to Formation of Translocations in Anaplastic Large Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 5831–5836. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.; Ohgi, K.; Zhang, J.; Rose, D.; Fu, X.-D.; Glass, C.K.; et al. Nuclear Receptor-Induced Chromosomal Proximity and DNA Breaks Underlie Specific Translocations in Cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Janz, S.; Potter, M.; Rabkin, C.S. Lymphoma- and Leukemia-Associated Chromosomal Translocations in Healthy Individuals. Genes Chromosomes Cancer 2003, 36, 211–223. [Google Scholar] [CrossRef]
- Rabkin, C.S.; Hirt, C.; Janz, S.; Dölken, G. T(14;18) Translocations and Risk of Follicular Lymphoma. J. Natl. Cancer Inst. Monogr. 2008, 39, 48–51. [Google Scholar] [CrossRef]
- Schüler, F.; Hirt, C.; Dölken, G. Chromosomal Translocation t(14;18) in Healthy Individuals. Semin. Cancer Biol. 2003, 13, 203–209. [Google Scholar] [CrossRef]
- Bäsecke, J.; Griesinger, F.; Trümper, L.; Brittinger, G. Leukemia- and Lymphoma-Associated Genetic Aberrations in Healthy Individuals. Ann. Hematol. 2002, 81, 64–75. [Google Scholar] [CrossRef]
- Brassesco, M.S.; Montaldi, A.P.; Gras, D.E.; de Paula Queiroz, R.G.; Martinez-Rossi, N.M.; Tone, L.G.; Sakamoto-Hojo, E.T. MLL Leukemia-Associated Rearrangements in Peripheral Blood Lymphocytes from Healthy Individuals. Genet. Mol. Biol. 2009, 32, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambiar, M.; Raghavan, S.C. Chromosomal Translocations among the Healthy Human Population: Implications in Oncogenesis. Cell Mol. Life Sci. 2013, 70, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb Perspect Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb Perspect Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Boboila, C.; Alt, F.W.; Schwer, B. Chapter One—Classical and Alternative End-Joining Pathways for Repair of Lymphocyte-Specific and General DNA Double-Strand Breaks. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 116, pp. 1–49. [Google Scholar]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic Homologous Recombination Maintains Genomic Stability and Suppresses Tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Brunet, E.; Jasin, M. Induction of Chromosomal Translocations with CRISPR-Cas9 and Other Nucleases: Understanding the Repair Mechanisms That Give Rise to Translocations. In Chromosome Translocation; Zhang, Y., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 15–25. ISBN 9789811305931. [Google Scholar]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol. Cell. Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef] [Green Version]
- Weinstock, D.M.; Richardson, C.A.; Elliott, B.; Jasin, M. Modeling Oncogenic Translocations: Distinct Roles for Double-Strand Break Repair Pathways in Translocation Formation in Mammalian Cells. DNA Repair 2006, 5, 1065–1074. [Google Scholar] [CrossRef]
- Ghosh, R.; Das, D.; Franco, S. The Role for the DSB Response Pathway in Regulating Chromosome Translocations. In Chromosome Translocation; Zhang, Y., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 65–87. ISBN 9789811305931. [Google Scholar]
- Kass, E.M.; Jasin, M. Collaboration and Competition between DNA Double-Strand Break Repair Pathways. FEBS Lett. 2010, 584, 3703–3708. [Google Scholar] [CrossRef] [Green Version]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-Induced Replication Repair of Damaged Forks Induces Genomic Duplications in Human Cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.D.; Jasin, M. Sister Chromatid Gene Conversion Is a Prominent Double-Strand Break Repair Pathway in Mammalian Cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Richardson, C.; Jasin, M. Coupled Homologous and Nonhomologous Repair of a Double-Strand Break Preserves Genomic Integrity in Mammalian Cells. Mol. Cell. Biol. 2000, 20, 9068–9075. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked Contribution of Alternative End-Joining to Chromosome-Translocation-Formation by Stochastically Induced DNA Double-Strand-Breaks in G2-Phase Human Cells. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2015, 793, 2–8. [Google Scholar] [CrossRef]
- Simsek, D.; Jasin, M. Alternative End-Joining Is Suppressed by the Canonical NHEJ Component Xrcc4-Ligase IV during Chromosomal Translocation Formation. Available online: https://pubmed.ncbi.nlm.nih.gov/20208544/ (accessed on 29 November 2020).
- Zhang, Y.; Jasin, M. An Essential Role for CtIP in Chromosomal Translocation Formation through an Alternative End-Joining Pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef]
- Boboila, C.; Jankovic, M.; Yan, C.T.; Wang, J.H.; Wesemann, D.R.; Zhang, T.; Fazeli, A.; Feldman, L.; Nussenzweig, A.; Nussenzweig, M.; et al. Alternative End-Joining Catalyzes Robust IgH Locus Deletions and Translocations in the Combined Absence of Ligase 4 and Ku70. Proc. Natl. Acad. Sci. USA 2010, 107, 3034–3039. [Google Scholar] [CrossRef] [Green Version]
- Elliott, B.; Richardson, C.; Jasin, M. Chromosomal Translocation Mechanisms at Intronic Alu Elements in Mammalian Cells. Mol. Cell 2005, 17, 885–894. [Google Scholar] [CrossRef]
- Löbrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative End-Joining Repair Pathways Are the Ultimate Backup for Abrogated Classical Non-Homologous End-Joining and Homologous Recombination Repair: Implications for the Formation of Chromosome Translocations. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhász, S.; Künzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA Double-Strand Break Resection Occurs during Non-Homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis Promote Homologous Recombination of Radiation-Induced DNA Double-Strand Breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB Repair Pathway Choice: An Orchestrated Handover Mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors Determining DNA Double-Strand Break Repair Pathway Choice in G2 Phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.-H.; Yoon, S.; Koh, Y.E.; Seo, Y.-J.; Kim, K.P. Maintenance of Genome Integrity and Active Homologous Recombination in Embryonic Stem Cells. Exp. Mol. Med. 2020, 52, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Mujoo, K.; Pandita, R.K.; Tiwari, A.; Charaka, V.; Chakraborty, S.; Singh, D.K.; Hambarde, S.; Hittelman, W.N.; Horikoshi, N.; Hunt, C.R.; et al. Differentiation of Human Induced Pluripotent or Embryonic Stem Cells Decreases the DNA Damage Repair by Homologous Recombination. Stem Cell Rep. 2017, 9, 1660–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukášová, E.; Kořistek, Z.; Klabusay, M.; Ondřej, V.; Grigoryev, S.; Bačíková, A.; Řezáčová, M.; Falk, M.; Vávrová, J.; Kohútová, V.; et al. Granulocyte Maturation Determines Ability to Release Chromatin NETs and Loss of DNA Damage Response; These Properties Are Absent in Immature AML Granulocytes. Biochim. Biophys. Acta 2013, 1833, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and Metabolism. Hypoxia, DNA Repair and Genetic Instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Lee, J.Y.; Orr-Weaver, T.L. Chromatin. In Encyclopedia of Genetics; Brenner, S., Miller, J.H., Eds.; Academic Press: New York, NY, USA, 2001; pp. 340–343. ISBN 978-0-12-227080-2. [Google Scholar]
- Thomas, J.O.; Kornberg, R.D. An Octamer of Histones in Chromatin and Free in Solution. Proc. Natl. Acad. Sci. USA 1975, 72, 2626–2630. [Google Scholar] [CrossRef] [Green Version]
- Arents, G.; Burlingame, R.W.; Wang, B.C.; Love, W.E.; Moudrianakis, E.N. The Nucleosomal Core Histone Octamer at 3.1 A Resolution: A Tripartite Protein Assembly and a Left-Handed Superhelix. Proc. Natl. Acad. Sci. USA 1991, 88, 10148–10152. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Campos, E.I.; Reinberg, D. Histones: Annotating Chromatin. Annu. Rev. Genet. 2009, 43, 559–599. [Google Scholar] [CrossRef]
- Misteli, T. The Cell Biology of Genomes: Bringing the Double Helix to Life. Cell 2013, 152, 1209–1212. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. Discovery and Characterization of Chromatin States for Systematic Annotation of the Human Genome. Nature Biotechnology 2010, 28, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. Chromatin-State Discovery and Genome Annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Marshall, W.F.; Straight, A.; Marko, J.F.; Swedlow, J.; Dernburg, A.; Belmont, A.; Murray, A.W.; Agard, D.A.; Sedat, J.W. Interphase Chromosomes Undergo Constrained Diffusional Motion in Living Cells. Curr. Biol. 1997, 7, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Gothe, H.J.; Minneker, V.; Roukos, V. Dynamics of Double-Strand Breaks: Implications for the Formation of Chromosome Translocations. In Chromosome Translocation; Zhang, Y., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 27–38. ISBN 9789811305931. [Google Scholar]
- Heun, P.; Laroche, T.; Shimada, K.; Furrer, P.; Gasser, S.M. Chromosome Dynamics in the Yeast Interphase Nucleus. Science 2001, 294, 2181–2186. [Google Scholar] [CrossRef]
- Neumann, F.R.; Dion, V.; Gehlen, L.R.; Tsai-Pflugfelder, M.; Schmid, R.; Taddei, A.; Gasser, S.M. Targeted INO80 Enhances Subnuclear Chromatin Movement and Ectopic Homologous Recombination. Genes Dev. 2012, 26, 369–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.C.; Spakowitz, A.J.; Theriot, J.A. Nonthermal ATP-Dependent Fluctuations Contribute to the in Vivo Motion of Chromosomal Loci. Proc. Natl. Acad. Sci. USA 2012, 109, 7338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, P.M.; Borovski, T.; Stap, J.; Cijsouw, T.; ten Cate, R.; Medema, J.P.; Kanaar, R.; Franken, N.a.P.; Aten, J.A. Chromatin Mobility Is Increased at Sites of DNA Double-Strand Breaks. J. Cell Sci. 2012, 125, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Thomson, I.; Gilchrist, S.; Bickmore, W.A.; Chubb, J.R. The Radial Positioning of Chromatin Is Not Inherited through Mitosis but Is Established De Novo in Early G1. Curr. Biol. 2004, 14, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.; Belmont, A.S.; Sedat, J.W. Multiple Regimes of Constrained Chromosome Motion Are Regulated in the Interphase Drosophila Nucleus. Curr. Biol. 2001, 11, 1227–1239. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Schermelleh, L.; Cremer, M.; Tashiro, S.; Cremer, T. Chromosome Order in HeLa Cells Changes during Mitosis and Early G1, but Is Stably Maintained during Subsequent Interphase Stages. J. Cell Biol. 2003, 160, 685–697. [Google Scholar] [CrossRef]
- Wiesmeijer, K.; Krouwels, I.M.; Tanke, H.J.; Dirks, R.W. Chromatin Movement Visualized with Photoactivable GFP-Labeled Histone H4. Differentiation 2008, 76, 83–90. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Carpenter, A.E.; Fuchsova, B.; Johnson, T.; de Lanerolle, P.; Belmont, A.S. Long-Range Directional Movement of an Interphase Chromosome Site. Curr. Biol. 2006, 16, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Dundr, M.; Ospina, J.K.; Sung, M.-H.; John, S.; Upender, M.; Ried, T.; Hager, G.L.; Matera, A.G. Actin-Dependent Intranuclear Repositioning of an Active Gene Locus in Vivo. J. Cell Biol. 2007, 179, 1095–1103. [Google Scholar] [CrossRef]
- Tumbar, T.; Belmont, A.S. Interphase Movements of a DNA Chromosome Region Modulated by VP16 Transcriptional Activator. Nat. Cell Biol. 2001, 3, 134–139. [Google Scholar] [CrossRef]
- Spichal, M.; Brion, A.; Herbert, S.; Cournac, A.; Marbouty, M.; Zimmer, C.; Koszul, R.; Fabre, E. Evidence for a Dual Role of Actin in Regulating Chromosome Organization and Dynamics in Yeast. J. Cell Sci. 2016, 129, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Therizols, P.; Illingworth, R.S.; Courilleau, C.; Boyle, S.; Wood, A.J.; Bickmore, W.A. Chromatin Decondensation Is Sufficient to Alter Nuclear Organization in Embryonic Stem Cells. Science 2014, 346, 1238–1242. [Google Scholar] [CrossRef]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in Chromatin Structure and Mobility in Living Cells at Sites of DNA Double-Strand Breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move Outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Kim, J.-A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin Is Refractory to γ-H2AX Modification in Yeast and Mammals. J. Cell Biol. 2007, 178, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear Position Dictates DNA Repair Pathway Choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [Green Version]
- Rosin, L.F.; Crocker, O.; Isenhart, R.L.; Nguyen, S.C.; Xu, Z.; Joyce, E.F. Chromosome Territory Formation Attenuates the Translocation Potential of Cells. eLife 2019, 8, e49553. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. More than Just a Focus: The Chromatin Response to DNA Damage and Its Role in Genome Integrity Maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Kaye, J.A.; Melo, J.A.; Cheung, S.K.; Vaze, M.B.; Haber, J.E.; Toczyski, D.P. DNA Breaks Promote Genomic Instability by Impeding Proper Chromosome Segregation. Curr. Biol. 2004, 14, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Lobachev, K.; Vitriol, E.; Stemple, J.; Resnick, M.A.; Bloom, K. Chromosome Fragmentation after Induction of a Double-Strand Break Is an Active Process Prevented by the RMX Repair Complex. Curr. Biol. 2004, 14, 2107–2112. [Google Scholar] [CrossRef] [Green Version]
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional Stability of Single Double-Strand Breaks in Mammalian Cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Lisby, M.; Mortensen, U.H.; Rothstein, R. Colocalization of Multiple DNA Double-Strand Breaks at a Single Rad52 Repair Centre. Nat. Cell Biol. 2003, 5, 572–577. [Google Scholar] [CrossRef]
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-Scholz, G. Live Cell Microscopy Analysis of Radiation-Induced DNA Double-Strand Break Motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177. [Google Scholar] [CrossRef] [Green Version]
- Caron, P.; Choudjaye, J.; Clouaire, T.; Bugler, B.; Daburon, V.; Aguirrebengoa, M.; Mangeat, T.; Iacovoni, J.S.; Álvarez-Quilón, A.; Cortés-Ledesma, F.; et al. Non-Redundant Functions of ATM and DNA-PKcs in Response to DNA Double-Strand Breaks. Cell Rep. 2015, 13, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Iarovaia, O.V.; Minina, E.P.; Sheval, E.V.; Onichtchouk, D.; Dokudovskaya, S.; Razin, S.V.; Vassetzky, Y.S. Nucleolus: A Central Hub for Nuclear Functions. Trends Cell Biol. 2019, 29, 647–659. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Shukla, A.; Grady, J.P.; Fink, J.L.; Dray, E.; Duijf, P.H.G. Translocation Breakpoints Preferentially Occur in Euchromatin and Acrocentric Chromosomes. Cancers (Basel) 2018, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuhara, T.; Xing, Y.-H.; Bauer, N.C.; Lee, L.; Dong, R.; Yadav, T.; Soberman, R.J.; Rivera, M.N.; Zou, L. Condensates Induced by Transcription Inhibition Localize Active Chromatin to Nucleoli. Mol. Cell 2022, 82, 2738–2753.e6. [Google Scholar] [CrossRef]
- Ramanand, S.G.; Mani, R.S. Stress and the CITI. Mol. Cell 2022, 82, 2730–2731. [Google Scholar] [CrossRef]
- Nelms, B.E.; Maser, R.S.; MacKay, J.F.; Lagally, M.G.; Petrini, J.H.J. In Situ Visualization of DNA Double-Strand Break Repair in Human Fibroblasts. Science 1998, 280, 590–592. [Google Scholar] [CrossRef] [Green Version]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased Mobility of Double-Strand Breaks Requires Mec1, Rad9 and the Homologous Recombination Machinery. Nat. Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef]
- Dion, V.; Kalck, V.; Seeber, A.; Schleker, T.; Gasser, S.M. Cohesin and the Nucleolus Constrain the Mobility of Spontaneous Repair Foci. EMBO Rep. 2013, 14, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Miné-Hattab, J.; Rothstein, R. Increased Chromosome Mobility Facilitates Homology Search during Recombination. Nat. Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Saad, H.; Gallardo, F.; Dalvai, M.; Tanguy-le-Gac, N.; Lane, D.; Bystricky, K. DNA Dynamics during Early Double-Strand Break Processing Revealed by Non-Intrusive Imaging of Living Cells. PLoS Genet. 2014, 10, e1004187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, A.L.; Kowalczykowski, S.C. Single-Molecule Imaging of DNA Pairing by RecA Reveals a Three-Dimensional Homology Search. Nature 2012, 482, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Ragunathan, K.; Liu, C.; Ha, T. RecA Filament Sliding on DNA Facilitates Homology Search. eLife 2012, 1, e00067. [Google Scholar] [CrossRef]
- Renkawitz, J.; Lademann, C.A.; Kalocsay, M.; Jentsch, S. Monitoring Homology Search during DNA Double-Strand Break Repair In Vivo. Mol. Cell 2013, 50, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Seeber, A.; Dion, V.; Gasser, S.M. Checkpoint Kinases and the INO80 Nucleosome Remodeling Complex Enhance Global Chromatin Mobility in Response to DNA Damage. Genes Dev. 2013, 27, 1999–2008. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA Repair by Nonhomologous End Joining and Homologous Recombination during Cell Cycle in Human Cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canoy, R.J.; Shmakova, A.; Karpukhina, A.; Shepelev, M.; Germini, D.; Vassetzky, Y. Factors That Affect the Formation of Chromosomal Translocations in Cells. Cancers 2022, 14, 5110. https://doi.org/10.3390/cancers14205110
Canoy RJ, Shmakova A, Karpukhina A, Shepelev M, Germini D, Vassetzky Y. Factors That Affect the Formation of Chromosomal Translocations in Cells. Cancers. 2022; 14(20):5110. https://doi.org/10.3390/cancers14205110
Chicago/Turabian StyleCanoy, Reynand Jay, Anna Shmakova, Anna Karpukhina, Mikhail Shepelev, Diego Germini, and Yegor Vassetzky. 2022. "Factors That Affect the Formation of Chromosomal Translocations in Cells" Cancers 14, no. 20: 5110. https://doi.org/10.3390/cancers14205110