
Metabolomic and Lipidomic Profiling of Gliomas—A New Direction in Personalized Therapies

Abstract
:Simple Summary
Abstract
1. Introduction
2. Metabolic Changes in Gliomas
Metabolomics and Lipidomics
3. Challenges in Glioma Therapy—Blood-Brain Barrier
4. Gold Standard Therapy
5. Immune Therapy—Monoclonal Antibodies
6. IDH Inhibitors and Epigenetic Therapies for Treatment IDH-Mutant Glioma
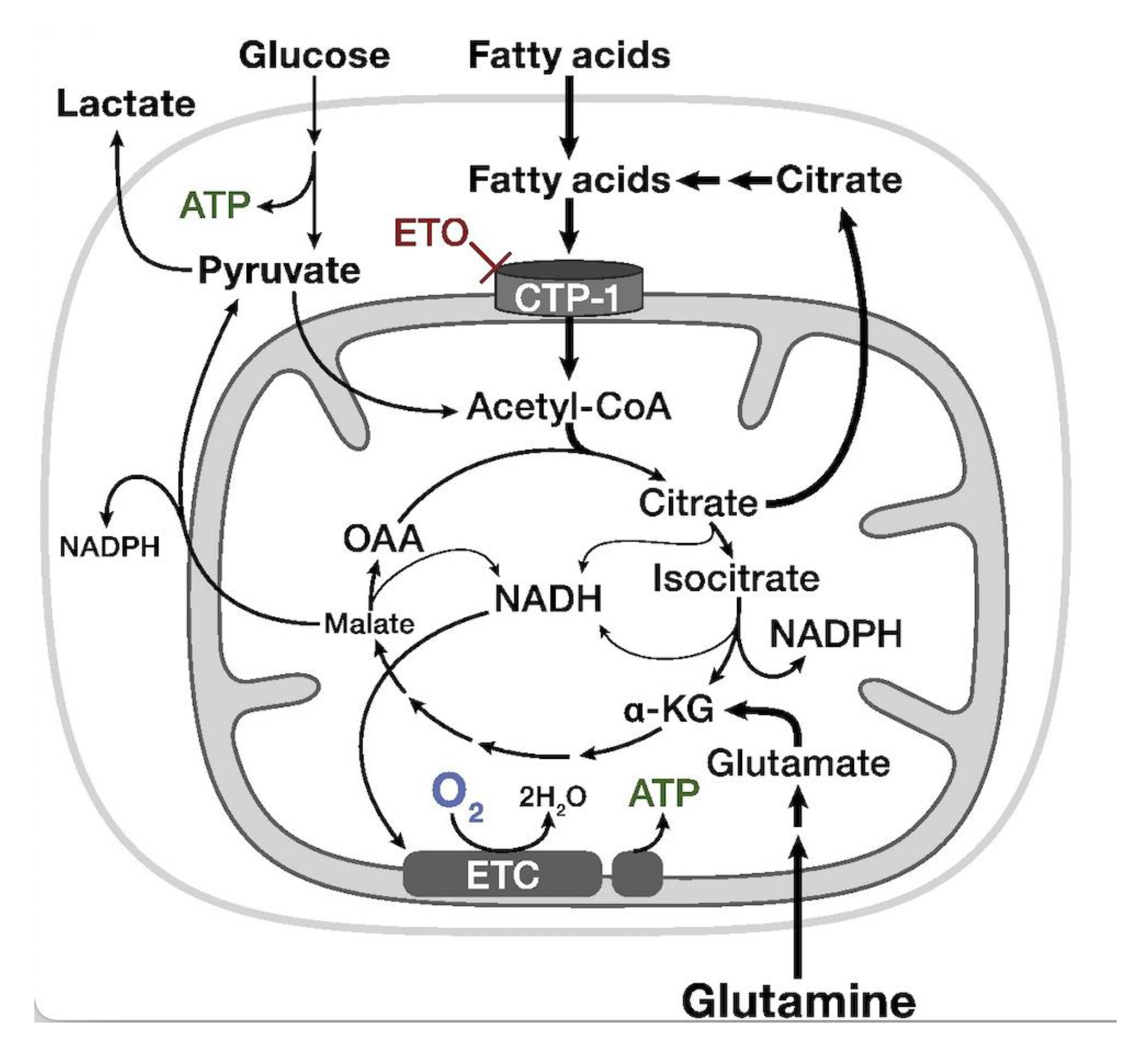
7. Carnitine Palmitoyl Transferase-1a (CPT-1a)-Inhibitor—Etomoxir
8. Topoisomerase-II Inhibitors
9. Other Approaches
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, M.E. Epidemiology and Overview of Gliomas. Semin. Oncol. Nurs. 2018, 34, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of Gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Frenkel, E.P.; Neuwelt, E.A. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology 2010, 76, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; Chapter 11; pp. 197–241. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic Signature of Brain Cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-Localized Biopsies Reveal Subtype-Specific Differences in Molecular and Cellular Composition at the Margins of Glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef] [Green Version]
- Uribe, D.; Niechi, I.; Rackov, G.; Erices, J.I.; Martín, R.S.; Quezada, C. Adapt to Persist: Glioblastoma Microenvironment and Epigenetic Regulation on Cell Plasticity. Biology 2022, 11, 313. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Juraszek, B.; Czarnecka-Herok, J.; Nałęcz, K.A. Glioma cells survival depends both on fatty acid oxidation and on functional carnitine transport by SLC22A5. J. Neurochem. 2020, 156, 642–657. [Google Scholar] [CrossRef]
- Bogusiewicz, J.; Goryńska, P.Z.; Gaca, M.; Chmara, K.; Goryński, K.; Jaroch, K.; Paczkowski, D.; Furtak, J.; Harat, M.; Bojko, B. On-Site Sampling and Extraction of Brain Tumors for Metabolomics and Lipidomics Analysis. J. Vis. Exp. 2020, 2020, e61260. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Jones, A.B.; Gordon, E.R.; Williford, S.E.; Harsh, Y.; Ziebro, J.K.; Landis, C.J.; Gc, S.; Crossman, D.K.; Cooper, S.J.; et al. Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling. Cells 2022, 11, 1873. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Mehta, A.; Awah, C.U.; Sonabend, A.M. Topoisomerase II Poisons for Glioblastoma; Existing Challenges and Opportunities to Personalize Therapy. Front. Neurol. 2018, 9, 459. [Google Scholar] [CrossRef]
- Carmen Lafita-Navarro, M.; Venkateswaran, N.; Kilgore, J.A.; Kanji, S.; Han, J.; Barnes, S.; Williams, N.S.; Buszczak, M.; Burma, S.; Conacci-Sorrell, M. Inhibition of the de Novo Pyrimidine Biosynthesis Pathway Limits Ribosomal RNA Transcription Causing Nucleolar Stress in Glioblastoma Cells. PLoS Genet. 2020, 16, e1009117. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kaplan, J.P.; Nguyen, H.; Stopka, S.A.; Savani, M.R.; Regan, M.S.; Nguyen, Q.-D.; Jones, K.L.; Moreau, L.A.; Peng, J.; et al. A Druggable Addiction to de Novo Pyrimidine Biosynthesis in Diffuse Midline Glioma. Cancer Cell 2022, 40, 957–972. [Google Scholar] [CrossRef]
- Shi, D.D.; Savani, M.R.; Levitt, M.M.; Wang, A.C.; Endress, J.E.; Bird, C.E.; Buehler, J.; Stopka, S.A.; Regan, M.S.; Lin, Y.-F.; et al. De Novo Pyrimidine Synthesis Is a Targetable Vulnerability in IDH Mutant Glioma. Cancer Cell 2022, 40, 939–956.e16. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, L.; Liang, J.; Huang, Q.; Sun, H. Neurotransmitters: Potential Targets in Glioblastoma. Cancers 2022, 14, 3970. [Google Scholar] [CrossRef]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schürer, S.C.; Ayad, N.G. Drug Repositioning in Glioblastoma: A Pathway Perspective. Front. Pharmacol. 2018, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Lehtimäki, K.K.; Valonen, P.K.; Griffin, J.L.; Väisänen, T.H.; Gröhn, O.H.J.; Kettunen, M.I.; Vepsäläinen, J.; Ylä-Herttuala, S.; Nicholson, J.; Kauppinen, R.A. Metabolite Changes in BT4C Rat Gliomas Undergoing Ganciclovir-Thymidine Kinase Gene Therapy-Induced Programmed Cell Death as Studied by 1H NMR Spectroscopy in Vivo, Ex Vivo, and in Vitro. J. Biol. Chem. 2003, 278, 45915–45923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, J.M.; Karve, A.S.; Gudelsky, G.A.; Gawali, M.V.; Seibel, W.; Sallans, L.; DasGupta, B.; Desai, P.B. Brain Pharmacokinetics and Metabolism of the AMP-Activated Protein Kinase Selective Inhibitor SBI-0206965, an Investigational Agent for the Treatment of Glioblastoma. Investig. New Drugs 2022, 40, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Omeljaniuk, W.J.; Krętowski, R.; Ratajczak-Wrona, W.; Jabłońska, E.; Cechowska-Pasko, M. Novel Dual Pi3k/Mtor Inhibitor, Apitolisib (Gdc-0980), Inhibits Growth and Induces Apoptosis in Human Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 11511. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; vander Heiden, M.G.; Locasale, J.W. Metabolomics in Cancer Research and Emerging Applications in Clinical Oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Duffy, M.J. Use of Biomarkers in Screening for Cancer. EJIFCC 2010, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Belhaj, M.R.; Lawler, N.G.; Hoffman, N.J. Metabolomics and Lipidomics: Expanding the Molecular Landscape of Exercise Biology. Metabolites 2021, 11, 151. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The Apogee of the Omics Trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.; Bernard, A.; Hau, A.; et al. Altered Metabolic Landscape in IDH -mutant Gliomas Affects Phospholipid, Energy, and Oxidative Stress Pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Miyata, S.; Tominaga, K.; Sakashita, E.; Urabe, M.; Onuki, Y.; Gomi, A.; Yamaguchi, T.; Mieno, M.; Mizukami, H.; Kume, A.; et al. Comprehensive Metabolomic Analysis of IDH1 R132H Clinical Glioma Samples Reveals Suppression of β-Oxidation Due to Carnitine Deficiency. Sci. Rep. 2019, 9, 9787. [Google Scholar] [CrossRef] [Green Version]
- Tap, W.D.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.J.; Jiang, L.; Wu, B.; et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients with Advanced Chondrosarcoma. J. Clin. Oncol. 2020, 38, 1693–1701. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-Mutant, Chemotherapy-Refractory Cholangiocarcinoma (ClarIDHy): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- St-Coeur, P.D.; Touaibia, M.; Cuperlovic-Culf, M.; Morin, P.J. Leveraging Metabolomics to Assess the next Generation of Temozolomide-Based Therapeutic Approaches for Glioblastomas. Genom. Proteom. Bioinform. 2013, 11, 199–206. [Google Scholar] [CrossRef]
- Goryńska, P.Z.; Chmara, K.; Kupcewicz, B.; Goryński, K.; Jaroch, K.; Paczkowski, D.; Furtak, J.; Harat, M.; Bojko, B. Metabolomic Phenotyping of Gliomas: What Can We Get with Simplified Protocol for Intact Tissue Analysis? Cancers 2022, 14, 312. [Google Scholar] [CrossRef]
- Pienkowski, T.; Kowalczyk, T.; Garcia-Romero, N.; Ayuso-Sacido, A.; Ciborowski, M. Proteomics and Metabolomics Approach in Adult and Pediatric Glioma Diagnostics. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188721. [Google Scholar] [CrossRef]
- Everett, J.R. From Metabonomics to Pharmacometabonomics: The Role of Metabolic Profiling in Personalized Medicine. Front. Pharmacol. 2016, 7, 297. [Google Scholar] [CrossRef]
- Kantae, V.; Krekels, E.H.J.; Esdonk, M.J.V.; Lindenburg, P.; Harms, A.C.; Knibbe, C.A.J.; van der Graaf, P.H.; Hankemeier, T. Integration of Pharmacometabolomics with Pharmacokinetics and Pharmacodynamics: Towards Personalized Drug Therapy. Metabolomics 2017, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, J.; Fan, Y.; Huang, T.; Zhou, Y.; Fan, H.; Zhang, Q.; Qiu, R. The Mechanism of Formononetin/Calycosin Compound Optimizing the Effects of Temozolomide on C6 Malignant Glioma Based on Metabolomics and Network Pharmacology. Biomed. Pharmacother. 2022, 153, 113418. [Google Scholar] [CrossRef]
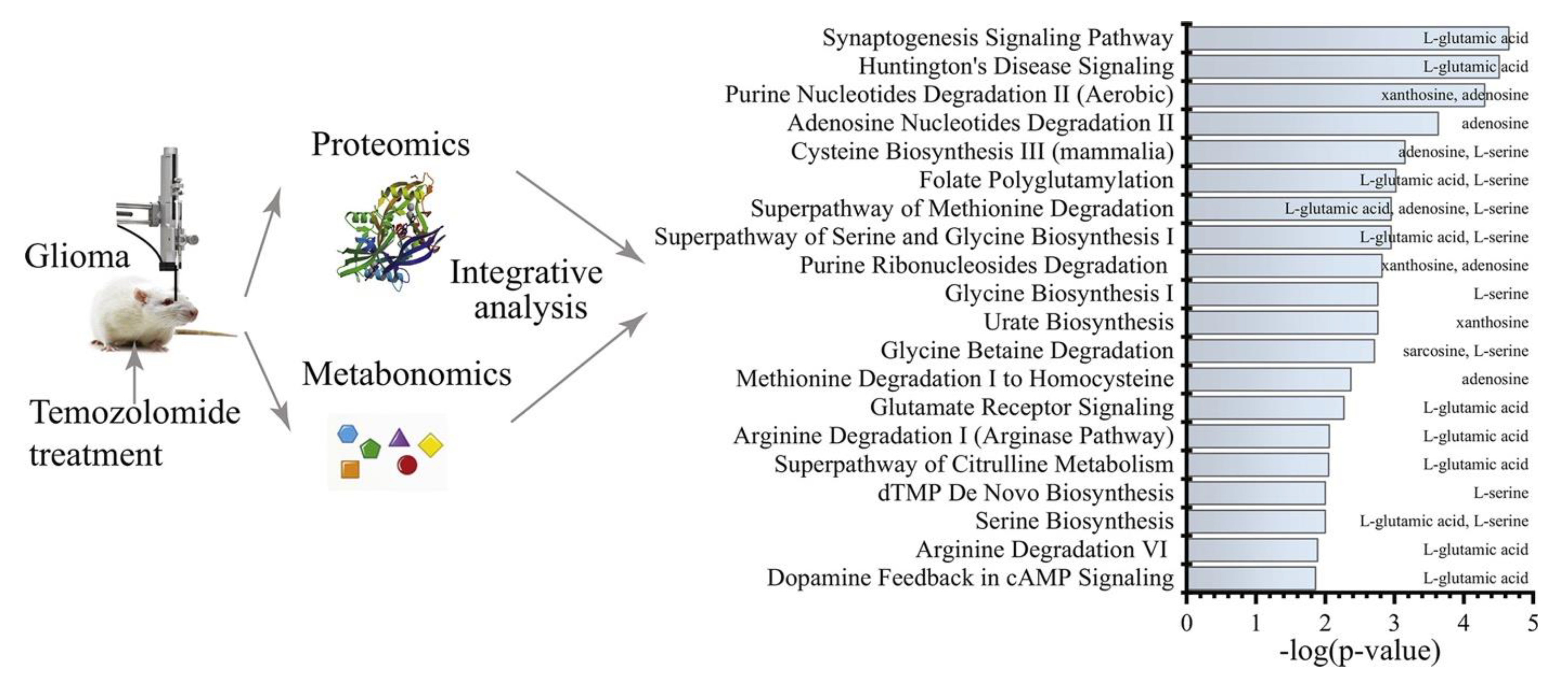
- Li, M.; Ren, T.; Lin, M.; Wang, Z.; Zhang, J. Integrated Proteomic and Metabolomic Profiling the Global Response of Rat Glioma Model by Temozolomide Treatment. J. Proteom. 2020, 211, 103578. [Google Scholar] [CrossRef]
- Voss, M.; Wagner, M.; von Mettenheim, N.; Harter, P.N.; Wenger, K.J.; Franz, K.; Bojunga, J.; Vetter, M.; Gerlach, R.; Glatzel, M.; et al. ERGO2: A Prospective, Randomized Trial of Calorie-Restricted Ketogenic Diet and Fasting in Addition to Reirradiation for Malignant Glioma. Int. J. Radiat. Oncol. 2020, 108, 987–995. [Google Scholar] [CrossRef]
- Valtorta, S.; lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and Temozolomide, a Synergic Option to Overcome Resistance in Glioblastoma Multiforme Models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [Green Version]
- St-Coeur, P.D.; Poitras, J.J.; Cuperlovic-Culf, M.; Touaibia, M.; Morin, P.J. Investigating a Signature of Temozolomide Resistance in GBM Cell Lines Using Metabolomics. J. Neurooncol. 2015, 125, 91–102. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Lo, W.L.; Chen, P.Y.; Ko, C.Y.; Chuang, J.Y.; Kao, T.J.; Yang, W.B.; Chang, K.Y.; Hung, C.Y.; Kikkawa, U.; et al. Reprogramming of Arachidonate Metabolism Confers Temozolomide Resistance to Glioblastoma through Enhancing Mitochondrial Activity in Fatty Acid Oxidation. J. Biomed. Sci. 2022, 29, 1–17. [Google Scholar] [CrossRef]
- Immanuel, S.R.C.; Ghanate, A.D.; Parmar, D.S.; Yadav, R.; Uthup, R.; Panchagnula, V.; Raghunathan, A. Integrated Genetic and Metabolic Landscapes Predict Vulnerabilities of Temozolomide Resistant Glioblastoma Cells. NPJ Syst. Biol. Appl. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Tiek, D.M.; Rone, J.D.; Graham, G.T.; Pannkuk, E.L.; Haddad, B.R.; Riggins, R.B. Alterations in Cell Motility, Proliferation, and Metabolism in Novel Models of Acquired Temozolomide Resistant Glioblastoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Mesti, T.; Bouchemal, N.; Banissi, C.; Triba, M.N.; Marbeuf-Gueye, C.; Cemazar, M.; le Moyec, L.; Carpentier, A.F.; Savarin, P.; Ocvirk, J. Nuclear Magnetic Resonance Metabolic Fingerprint of Bevacizumab in Mutant IDH1 Glioma Cells. Radiol. Oncol. 2018, 52, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab Treatment Induces Metabolic Adaptation toward Anaerobic Metabolism in Glioblastomas. Acta Neuropathol. 2015, 129, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.H.; Liu, G.Y.; Wang, R.; Moon, S.H.; Gross, R.W.; Patti, G.J. Identifying Off-Target Effects of Etomoxir Reveals That Carnitine Palmitoyltransferase i Is Essential for Cancer Cell Proliferation Independent of β-Oxidation. PLoS Biol. 2018, 16. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced Fatty Acid Oxidation Provides Glioblastoma Cells Metabolic Plasticity to Accommodate to Its Dynamic Nutrient Microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef]
- Nałęcz, K.A. Solute Carriers in the Blood–Brain Barier: Safety in Abundance. Neurochem. Res. 2017, 42, 795–809. [Google Scholar] [CrossRef]
- Da Ros, M.; de Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhairiyah, F.; de Lange, E.C.M. Understanding Drug Delivery to the Brain Using Liposome-Based Strategies: Studies That Provide Mechanistic Insights Are Essential. AAPS J. 2021, 23, 114. [Google Scholar] [CrossRef] [PubMed]
- Ananda, S.; Nowak, A.K.; Cher, L.; Dowling, A.; Brown, C.; Simes, J.; Rosenthal, M.A. Phase 2 Trial of Temozolomide and Pegylated Liposomal Doxorubicin in the Treatment of Patients with Glioblastoma Multiforme Following Concurrent Radiotherapy and Chemotherapy. J. Clin. Neurosci. 2011, 18, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hong, W.; Yu, M.; Li, Y.; Zheng, Y.; Ying, X. Multifunctional Targeting Liposomes of Epirubicin Plus Resveratrol Improved Therapeutic Effect on Brain Gliomas. Int. J. Nanomed. 2022, 17, 1087–1110. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Wesolowski, J.R.; Rajdev, P.; Mukherji, S.K. Temozolomide (Temodar). Am. J. Neuroradiol. 2010, 31, 1383–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galán-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT Genomic Rearrangements Contribute to Chemotherapy Resistance in Gliomas. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Sboarina, M.; Cacace, A.; Grasso, D.; Thabault, L.; Hamelin, L.; Vazeille, T.; Dumon, E.; Rossignol, R.; Frédérick, R.; et al. Olaparib Is a Mitochondrial Complex i Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 11938. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Miyake, K.; Yoshida, K.; Sasaki, H. Bevacizumab for Malignant Gliomas: Current Indications, Mechanisms of Action and Resistance, and Markers of Response. Brain Tumor Pathol. 2017, 34, 62–77. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Kim, M.M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma Past, Present, and Future Directions. Cancer J. 2018, 24, 180–186. [Google Scholar] [CrossRef]
- Lombardi, G.; Barresi, V.; Indraccolo, S.; Simbolo, M.; Fassan, M.; Mandruzzato, S.; Simonelli, M.; Caccese, M.; Pizzi, M.; Fassina, A.; et al. Pembrolizumab Activity in Recurrent High-Grade Gliomas with Partial or Complete Loss of Mismatch Repair Protein Expression: A Monocentric, Observational and Prospective Pilot Study. Cancers 2020, 12, 2283. [Google Scholar] [CrossRef]
- Govindarajan, V.; Shah, A.H.; Di, L.; Rivas, S.; Suter, R.K.; Eichberg, D.G.; Luther, E.; Lu, V.; Morell, A.A.; Ivan, M.E.; et al. Systematic Review of Epigenetic Therapies for Treatment of IDH-Mutant Glioma. World Neurosurg. 2022, 162, 47–56. [Google Scholar] [CrossRef]
- Pang, H.; Jia, W.; Hu, Z. Emerging Applications of Metabolomics in Clinical Pharmacology. Clin. Pharmacol. Ther. 2019, 106, 544–556. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of Mutant-IDH1 Inhibitors in Glioma Patients Probed by in Vivo 3D MRS Imaging of 2-Hydroxyglutarate. Nat. Commun. 2018, 9, 1474. [Google Scholar] [CrossRef] [Green Version]
- Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; De La Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in Isocitrate Dehydrogenase 1–Mutated Advanced Glioma. J. Clin. Oncol. 2020, 38, 3398–3406. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Penas-Prado, M.; Peters, K.B.; Burris, H.A.; Maher, E.A.; Janku, F.; Cote, G.M.; de la Fuente, M.I.; Clarke, J.L.; Ellingson, B.M.; et al. Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial. Clin. Cancer Res. 2021, 27, 4491–4499. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The Carnitine System and Cancer Metabolic Plasticity Review-Article. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Patel, S.; Affeck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty Acid Oxidation Is Required for the Respiration and Proliferation of Malignant Glioma Cells. Neuro-Oncology 2016, 19, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Vishwanath, V.A. Fatty Acid Beta-Oxidation Disorders: A Brief Review. Ann. Neurosci. 2016, 23, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of Fatty Acid Oxidation by Etomoxir Impairs NADPH Production and Increases Reactive Oxygen Species Resulting in ATP Depletion and Cell Death in Human Glioblastoma Cells. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A Novel Approach to Target Hypoxic Cancer Cells via Combining β-Oxidation Inhibitor Etomoxir with Radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid Accumulation and Oxidation in Glioblastoma Multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef] [Green Version]
- Trabjerg, M.S.; Mørkholt, A.S.; Lichota, J.; Oklinski, M.K.E.; Andersen, D.C.; Jønsson, K.; Mørk, K.; Skjønnemand, M.L.N.; Kroese, L.J.; Pritchard, C.E.J.; et al. Dysregulation of Metabolic Pathways by Carnitine Palmitoyl-Transferase 1 Plays a Key Role in Central Nervous System Disorders: Experimental Evidence Based on Animal Models. Sci. Rep. 2020, 10, 15583. [Google Scholar] [CrossRef]
- Lee, J.H.; Mosher, E.P.; Lee, Y.S.; Bumpus, N.N.; Berger, J.M. Control of Topoisomerase II Activity and Chemotherapeutic Inhibition by TCA Cycle Metabolites. Cell Chem. Biol. 2021, 29, 476–489.e6. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Sampson, J.H.; Sathornsumetee, S.; McLendon, R.E.; Herndon, J.E.; Marcello, J.E.; Norfleet, J.; et al. Metronomic Chemotherapy with Daily, Oral Etoposide plus Bevacizumab for Recurrent Malignant Glioma: A Phase II Study. Br. J. Cancer 2009, 101, 1986–1994. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, Y.; Hou, X.; Gao, X.; Shi, Q.; Li, S.; Huang, H. Combined Carboplatin and Etoposide Chemotherapy for Patients with Recurrent Glioma. Ann. Palliat. Med. 2021, 10, 12650–12656. [Google Scholar] [CrossRef]
- Perry, J.R.; Bélanger, K.; Mason, W.P.; Fulton, D.; Kavan, P.; Easaw, J.; Shields, C.; Kirby, S.; Macdonald, D.R.; Eisenstat, D.D.; et al. Phase II Trial of Continuous Dose-Intense Temozolomide in Recurrent Malignant Glioma: RESCUE Study. J. Clin. Oncol. 2010, 28, 2051–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.J.; Upadhyayula, P.S.; Pouliopoulos, A.N.; Englander, Z.K.; Zhang, X.; Jan, C.I.; Guo, J.; Mela, A.; Zhang, Z.; Wang, T.J.C.; et al. Focused Ultrasound-Mediated Blood-Brain Barrier Opening Increases Delivery and Efficacy of Etoposide for Glioblastoma Treatment. Int. J. Radiat. Oncol. 2021, 110, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fusfoundation.org/posts/fda-approves-focused-ultrasound-treatment-for-parkinsons-disease/ (accessed on 10 June 2022).
- Fishman, P.S.; Frenkel, V. Focused Ultrasound: An Emerging Therapeutic Modality for Neurologic Disease. Neurotherapeutics 2017, 14, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonabend, A.M.; Carminucci, A.S.; Amendolara, B.; Bansal, M.; Leung, R.; Lei, L.; Realubit, R.; Li, H.; Karan, C.; Yun, J.; et al. Convection-Enhanced Delivery of Etoposide Is Effective against Murine Proneural Glioblastoma. Neuro-Oncology 2014, 16, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Goñi, T.; Julià-Sapé, M.; Candiota, A.P.; Pumarola, M.; Arús, C. Molecular Imaging Coupled to Pattern Recognition Distinguishes Response to Temozolomide in Preclinical Glioblastoma. NMR Biomed. 2014, 27, 1333–1345. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Goal | Studied Species | Biological Material | Analytical Platform | Significant Metabolites | Altered Metabolic Pathways | Reference |
|---|---|---|---|---|---|---|---|
| Temozolomide | The effect of the complex of formononetin and calycosin on glioma temozolomide sensitivity | Rat, injected with C6 cells | C6 cell lines | GC-MS | Treatment is related with
treatment compared to the group treated only with temozolomide. |
| [39] |
| Temozolomide | The effect of the complex of formononetin and calycosin on glioma temozolomide sensitivity | Rat, injected with C6 cells | Serum | GC-MS | Treatment is related with
treatment compared to the group treated only with temozolomide. |
| [39] |
| Temozolomide | The effect of the complex of formononetin and calycosin on glioma temozolomide sensitivity | Rat, injected with C6 cells | Glioma tissue | GC-MS | Treatment is related with:
treatment compared to the group treated only with temozolomide. |
| [39] |
| Temozolomide | Response to temozolomide treatment | Rat, injected with C6 cells | Plasma | LC-MS | Treatment is related with
|
| [40] |
| Temozolomide | Response to temozolomide treatment | Rat, injected with C6 cells | Tumor tissue | LC-MS | Treatment is related with
(without treatment). |
| [40] |
| Temozolomide | Response to temozolomide treatment | Mouse, injected with GL261 cells | Tumor tissue | NMR | Treatment using TMZ is related to changes in:
| … | [41] |
| Temozolomide | The effect of metformin on glioma temozolomide sensitivity | Mouse, injected with TMZ-sensitive U251 and TMZ-resistant T98G cells | Tumor tissue | GC-MS | The application of TMZ with metformin compared to treatment with TMZ alone:
|
| [42] |
| Temozolomide | Response to temozolomide treatment | --- | Cell lines (TMZ-sensitive and TMZ-resistant U373 cells) | NMR | Treatment using TMZ is related with:
Combination of TMZ with lomeguatrib is related with:
Independently to used treatment:
| Treatment using TMZ is related with:
| [43] |
| Temozolomide | TMZ-sensitive and TMZ-resistant glioblastoma profiling | Human | Brain tumor tissue | NMR | Treatment using TMZ is related with:
|
| [43] |
| Temozolomide | Response to temozolomide treatment | --- | Cell lines (U87) | LCMS | Treatment using TMZ is related with:
|
| [44] |
| Temozolomide | Profiling the metabolome of TMZ-sensitive and TMZ-resistant cells | --- | Cell lines (NSP—TMZ resistant; U87M) | LC-MS | Response to TMZ doses:
TMZ effect on intracellular metabolome:
TMZ effect on extracellular metabolome:
| Response to TMZ doses:
| [45] |
| Temozolomide | Profiling the metabolome of TMZ-sensitive and TMZ-resistant cells | --- | Cell lines (TMZ sensitive and resistant 8MBGA) medium | LC-MS, GC-MS | Treatment using TMZ is related with:
|
| [46] |
| Bevacizumab | Assessment of bevacizumab effectiveness | --- | Cell lines (mIDH1-U87) | NMR | Treatment is related with:
|
| [47] |
| Bevacizumab | Response to bevacizumab treatment | Human | Tumor tissue | LC-MS | Treatment is related with:
|
| [48] |
| Etomoxir | Assessment of etomoxir effectiveness | --- | Cell lines (BT549) | LC-MS | Treatment is related with:
|
| [49] |
| Etomoxir | Assessment of etomoxir effectiveness | --- | Cell lines (Mesenchymal (MES83, MES326, and MES1027A) and proneural (PN19, PN84)) | LCMS | Treatment is related with:
|
| [50] |
| Invasive Methods | Noninvasive Methods |
|---|---|
| Brain microdialysis | Prodrugs |
| Intracerebral implantation | BBB permeability modulation |
| Intraventricular delivery | Nanotechnologies |
| Receptor mediated transport |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaca-Tabaszewska, M.; Bogusiewicz, J.; Bojko, B. Metabolomic and Lipidomic Profiling of Gliomas—A New Direction in Personalized Therapies. Cancers 2022, 14, 5041. https://doi.org/10.3390/cancers14205041
Gaca-Tabaszewska M, Bogusiewicz J, Bojko B. Metabolomic and Lipidomic Profiling of Gliomas—A New Direction in Personalized Therapies. Cancers. 2022; 14(20):5041. https://doi.org/10.3390/cancers14205041
Chicago/Turabian StyleGaca-Tabaszewska, Magdalena, Joanna Bogusiewicz, and Barbara Bojko. 2022. "Metabolomic and Lipidomic Profiling of Gliomas—A New Direction in Personalized Therapies" Cancers 14, no. 20: 5041. https://doi.org/10.3390/cancers14205041