
A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data source and Processing
2.2. Candidate Selection and Signature Establishment
2.3. Quantitative RT-qPCR and Risk Score Calculations of Clinical Cohort
2.4. Bioinformatics and Statistical Analyses
3. Results
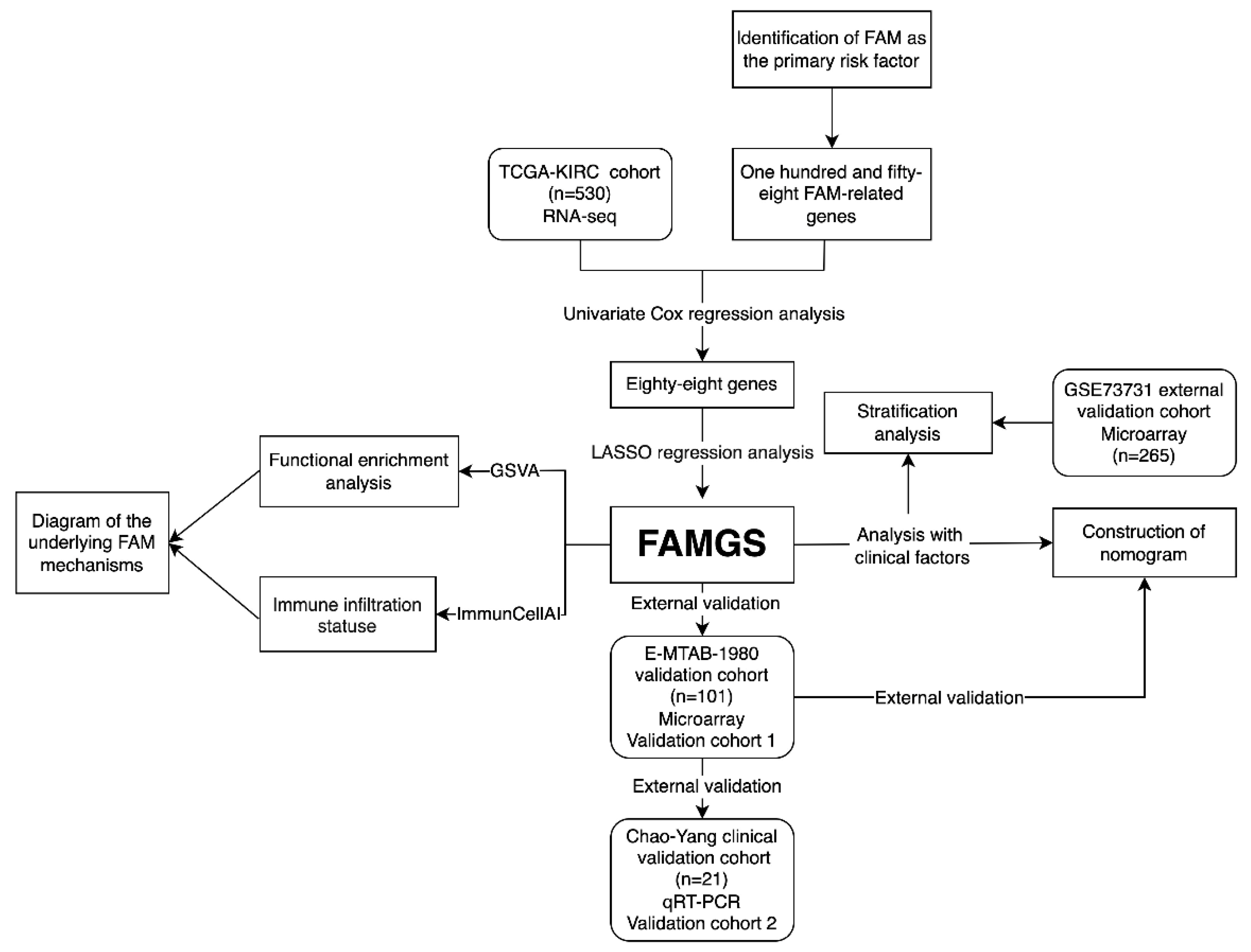
3.1. Study Design and Cohort Characteristics
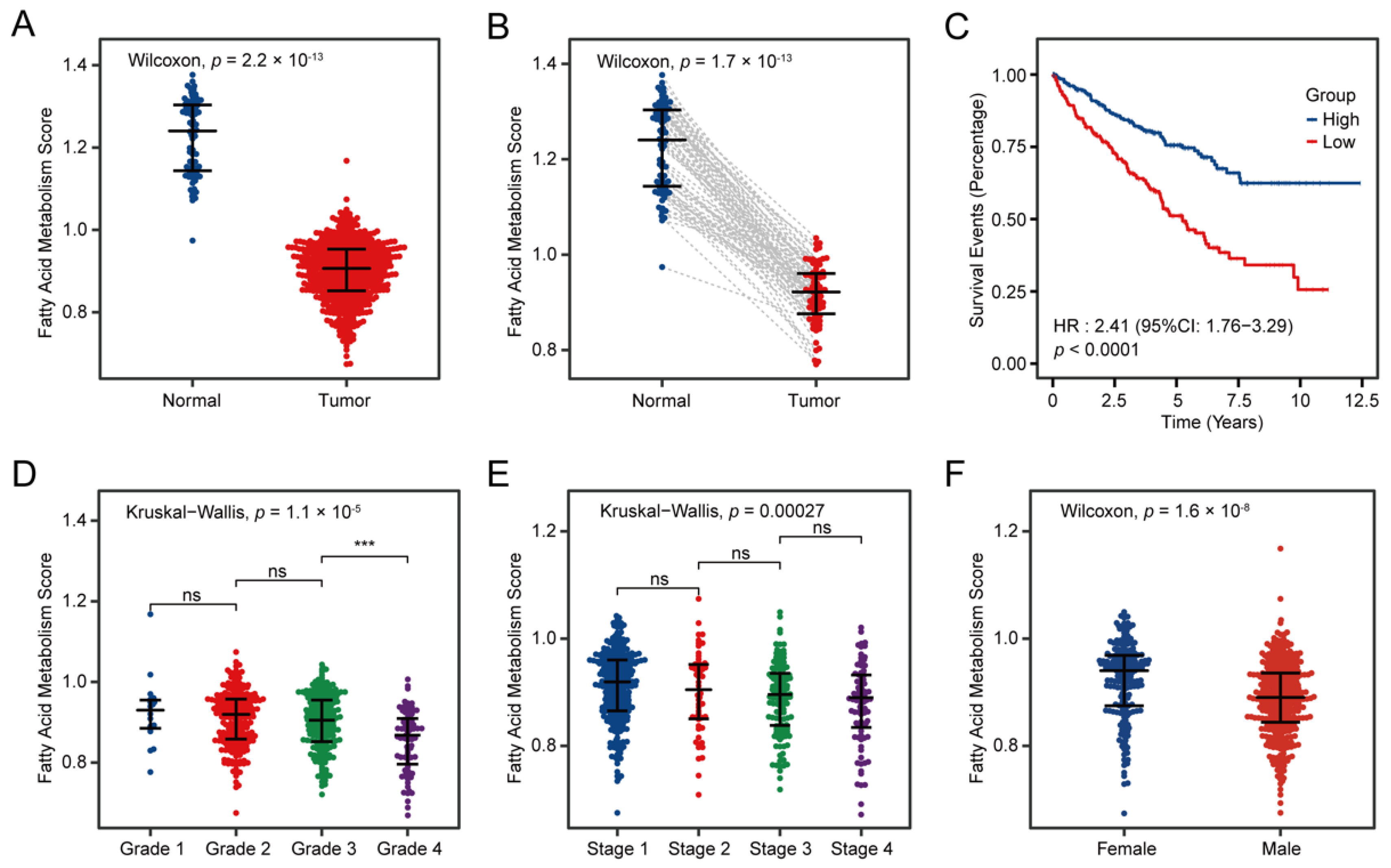
3.2. Fatty Acid Metabolism Confirmed as a Crucial Factor in ccRCC
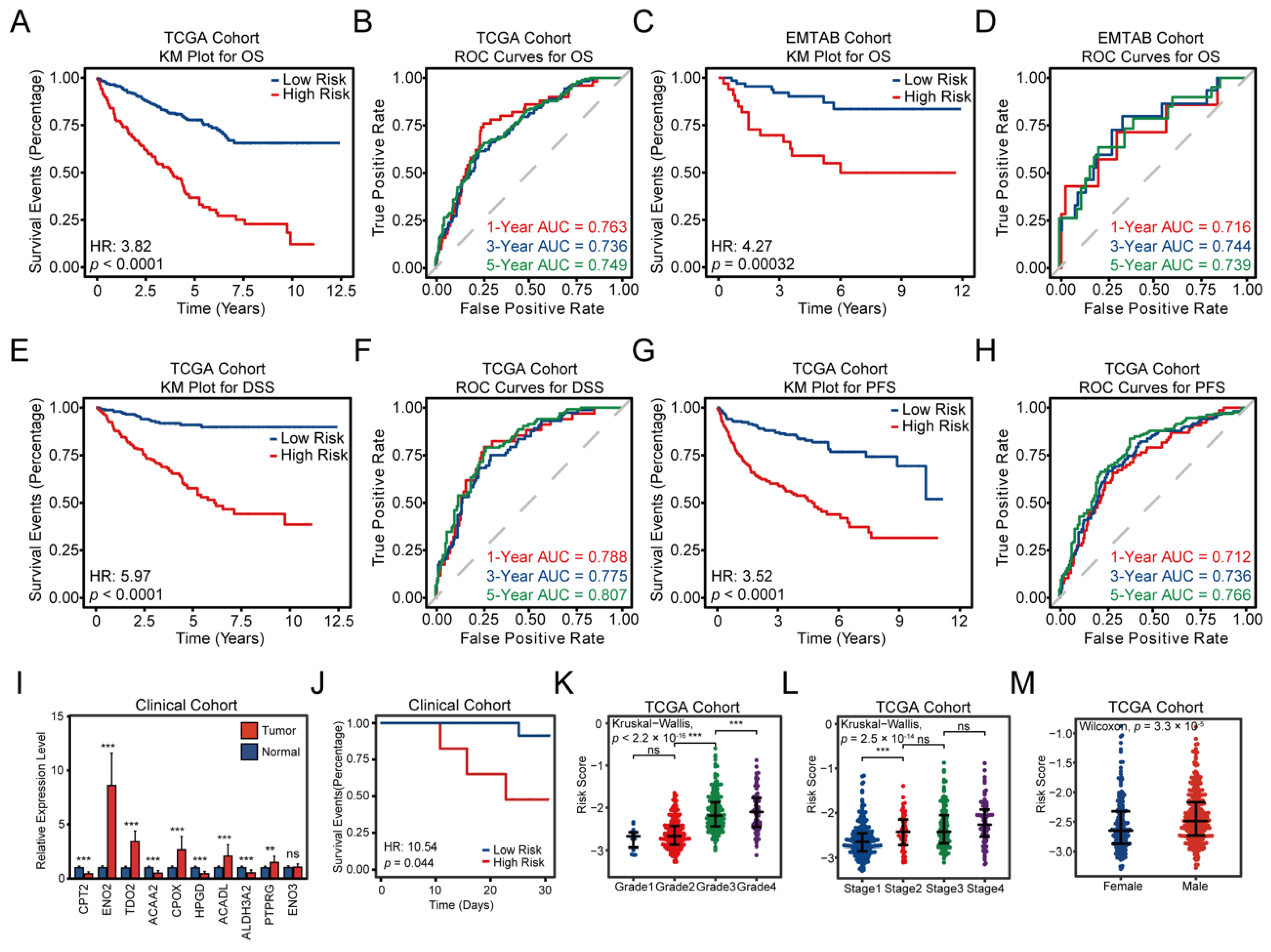
3.3. Construction and Validation of the FAMGS for Prognosis
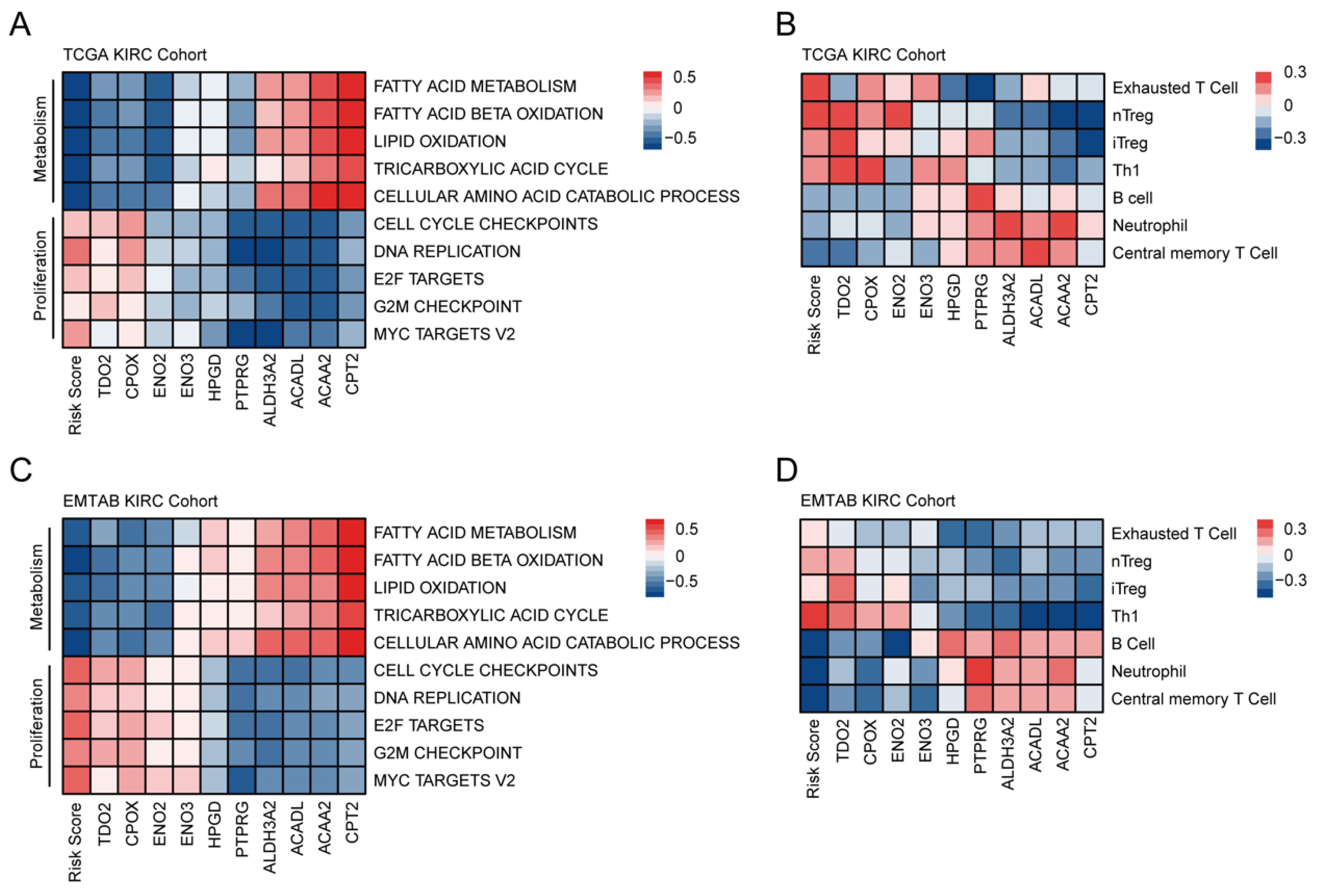
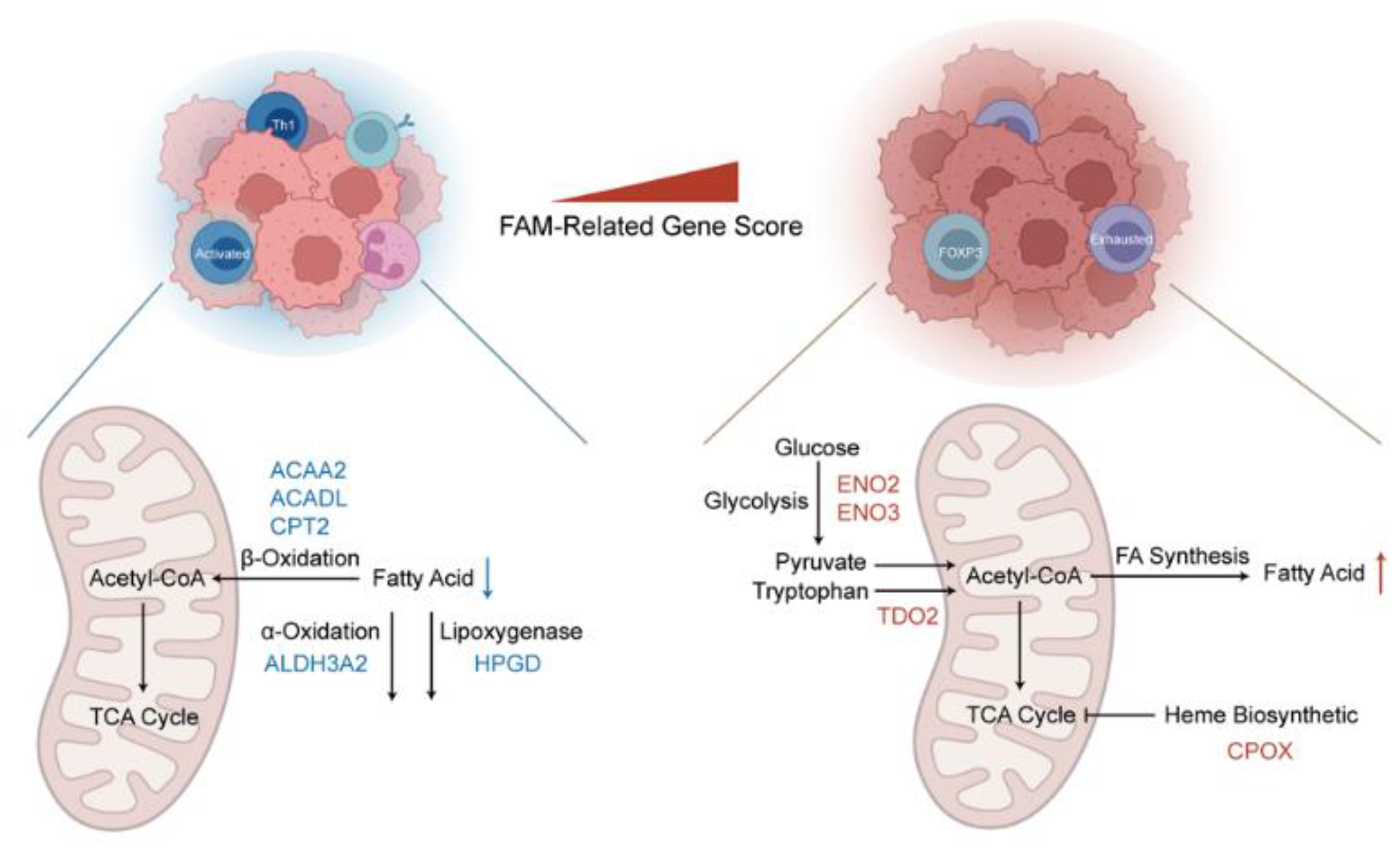
3.4. Comprehensive Enrichment Analyses and Immune Infiltration
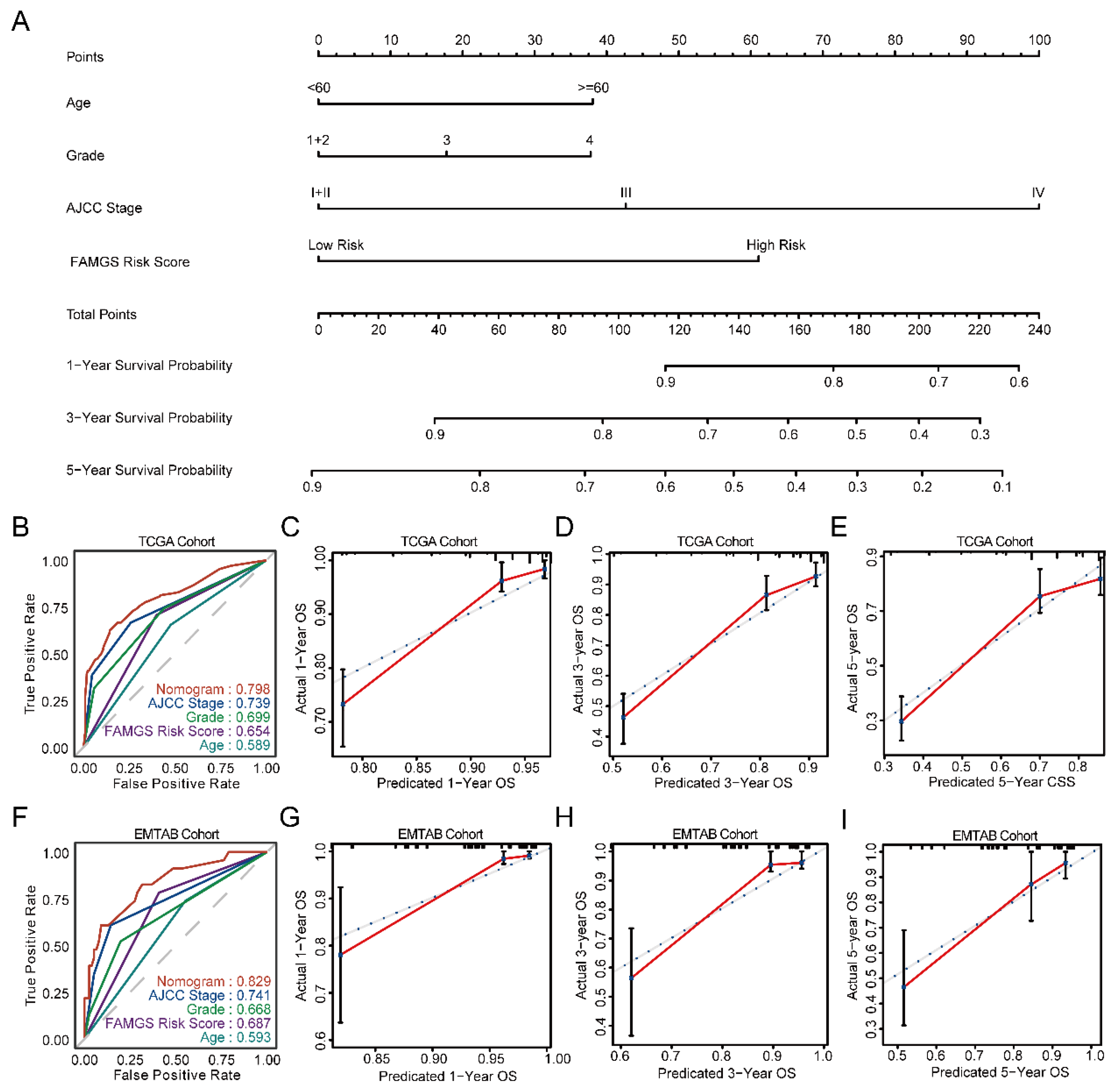
3.5. Establishment and Verification of a Nomogram Model According to the FAMGS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the Tumor Microenvironment: From Cancer Progression to Treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Brit. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty Acid Synthase over Expression Is an Indicator of Tumor Aggressiveness and Poor Prognosis in Renal Cell Carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N.; Weiss, R.H. Metabolic Reprogramming in Clear Cell Renal Cell Carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program-NCI. Available online: https://portal.gdc.cancer.gov (accessed on 24 July 2022).
- E-MTAB-1980 <Browse <ArrayExpress <EMBL-EBI. Available online: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1980/ (accessed on 24 July 2022).
- GEO Accession Viewer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73731 (accessed on 24 July 2022).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Tibshirani, R. The Lasso Method for Variable Selection in the Cox Model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Dyrskjøt, L.; Reinert, T.; Novoradovsky, A.; Zuiverloon, T.C.M.; Beukers, W.; Zwarthoff, E.; Malats, N.; Real, F.X.; Segersten, U.; Malmström, P.-U.; et al. Analysis of Molecular Intra-Patient Variation and Delineation of a Prognostic 12-Gene Signature in Non-Muscle Invasive Bladder Cancer; Technology Transfer from Microarrays to PCR. Br. J. Cancer 2012, 107, 1392–1398. [Google Scholar] [CrossRef]
- Dyrskjøt, L.; Reinert, T.; Algaba, F.; Christensen, E.; Nieboer, D.; Hermann, G.G.; Mogensen, K.; Beukers, W.; Marquez, M.; Segersten, U.; et al. Prognostic Impact of a 12-Gene Progression Score in Non–Muscle-Invasive Bladder Cancer: A Prospective Multicentre Validation Study. Eur. Urol. 2017, 72, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Lumley, T.; Elizabeth, A.; Cynthia, C. Survival: Survival Analysis. 2022. Available online: https://github.com/therneau/survival (accessed on 24 July 2022).
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-Dependent ROC Curves for Censored Survival Data and a Diagnostic Marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.-R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.-Y.; Wang, H.; Guo, A.-Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and Its Application in Cancer Immunotherapy. Adv. Sci. (Weinh) 2020, 7, 1902880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://github.com/raivokolde/pheatmap (accessed on 24 July 2022).
- Zhang, Z.; Kattan, M.W. Drawing Nomograms with R: Applications to Categorical Outcome and Survival Data. Ann. Transl. Med. 2017, 5, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljungberg, B.; Bensalah, K.; Canfield, S.; Dabestani, S.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.; Marconi, L.; Merseburger, A.S.; et al. EAU Guidelines on Renal Cell Carcinoma: 2014 Update. Eur. Urol. 2015, 67, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, X.; Li, Y.; Liu, Q.; Wu, F.; Qu, H.; Gao, H.; Ge, J.; Xu, Y.; Wang, H.; et al. Expression and Prognostic Significance of Fatty Acid Synthase in Clear Cell Renal Cell Carcinoma. Pathol.-Res. Pract. 2020, 216, 153227. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Cai, L.; Huang, W.; Weng, Q.; Lin, X.; You, M.; Liao, S. Prognostic Value of Fatty Acid Metabolism-Related Genes in Patients with Hepatocellular Carcinoma. Aging 2021, 13, 17847–17863. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Xu, C.; Wen, J.; Zhang, Y.; Wang, M.; Liu, X.; Zhao, K.; Wang, Z.; Liu, Y.; Zhang, T. Fatty Acid Metabolism-Related LncRNAs Are Potential Biomarkers for Predicting the Overall Survival of Patients With Colorectal Cancer. Front. Oncol. 2021, 11, 704038. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, D.; Lu, Q.; Yao, Y.; Ji, C. Bioinformatic Profiling Identifies a Fatty Acid Metabolism-Related Gene Risk Signature for Malignancy, Prognosis, and Immune Phenotype of Glioma. Dis. Markers 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Cheng, G.; Ye, Y.; Le, C.; Miao, Q.; Chen, J.; Yang, H.; Zhang, X. A Fatty Acid Metabolism Signature Associated With Clinical Therapy in Clear Cell Renal Cell Carcinoma. Front. Genet. 2022, 13, 18. [Google Scholar] [CrossRef]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear Cell Renal Cell Carcinoma Ontogeny and Mechanisms of Lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Z.; Bao, L.; Zhou, L.; Hou, Y.; Liu, L.; Xiong, M.; Zhang, Y.; Wang, B.; Tao, Z.; et al. Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in CcRCC, Which Results in Different Clinical Outcomes. Mol. Ther. 2020, 28, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A. Lipid Signalling Enforces Functional Specialization of Treg Cells in Tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef]
- González-Romero, F.; Mestre, D.; Aurrekoetxea, I.; O’Rourke, C.J.; Andersen, J.B.; Woodhoo, A.; Tamayo-Caro, M.; Varela-Rey, M.; Palomo-Irigoyen, M.; Gómez-Santos, B.; et al. E2F1 and E2F2-Mediated Repression of CPT2 Establishes a Lipid-Rich Tumor-Promoting Environment. Cancer Res. 2021, 81, 2874–2887. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Guo, D.; Zhang, X.; Zhu, Y.; Wang, Z.; Jin, Y.; Guo, W.; Zhang, S. ENO3 Inhibits Growth and Metastasis of Hepatocellular Carcinoma via Wnt/β-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 797102. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy–Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-Related Genes Serve as Potential Prognostic Biomarkers in Clear Cell Renal Cell Carcinoma. Oxidative Med. Cell. Longev. 2021, 2021, 6699808. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chu, W.; Yang, W.; Cheng, Y.; Chu, C.; Pan, X.; Ye, J.; Cao, J.; Gan, S.; Cui, X. Identification of RNA Transcript Makers Associated With Prognosis of Kidney Renal Clear Cell Carcinoma by a Competing Endogenous RNA Network Analysis. Front. Genet. 2020, 11, 540094. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, L.; Hu, Z.; Peng, D. Integrated Multi-Omics Analysis Identified PTPRG and CHL1 as Key Regulators of Immunophenotypes in Clear Cell Renal Cell Carcinoma(CcRCC). Front. Oncol. 2022, 12, 832027. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tao, Z.; Chen, X. A Three-Metabolic-Genes Risk Score Model Predicts Overall Survival in Clear Cell Renal Cell Carcinoma Patients. Front. Oncol. 2020, 10, 570281. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | TCGA Training Cohort | EMTAB Validation Cohort | Chao-Yang Validation Cohort | |
|---|---|---|---|---|
| Number of Patients | 530 | 101 | 21 | |
| Overall Survival (IQR) | 1181.5 (520, 1912) | 1530 (1020, 2430) | 848 (712,916) | |
| Overall Survival Status (%) | Survival | 357 (67.36) | 78 (77.23) | 17 (80.95%) |
| Deceased | 173 (32.64) | 23 (22.77) | 4 (19.05%) | |
| Age (IQR) | 61 (52, 70) | 64 (56, 72) | 67 (62.5, 72.5) | |
| Gender (%) | Male | 344 (64.91) | 77 (76.24) | 16 (76.19%) |
| Female | 186 (35.09) | 24 (23.76) | 5 (23.81%) | |
| Grade (%) | G1 | 14 (2.64) | 13 (12.87) | 2 (9.52%) |
| G2 | 227 (42.83) | 59 (58.42) | 14 (57.14%) | |
| G3 | 207 (39.06) | 22 (21.78) | 5 (23.81%) | |
| G4 | 74 (13.96) | 5 (4.95) | 0 (0.00) | |
| Not Available | 8 (1.51) | 2 (1.98) | 0 (0.00) | |
| AJCC Stage (%) | Stage I | 265 (50.00) | 66 (65.35) | 13 (61.91%) |
| Stage II | 57 (10.75) | 10 (9.90) | 4 (19.05%) | |
| Stage III | 123 (23.21) | 13 (12.87) | 2 (9.52%) | |
| Stage IV | 82 (15.47) | 12 (11.88) | 2 (9.52%) | |
| Not Available | 3 (0.57) | 0 (0.00) | 0 (0.00) |
| TCGA Training Cohort | ||||||
|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||
| Factors | HR (95% CI) | p Value | HR (95% CI) | p Value | ||
| FAMGS Risk Score | 3.729 (2.752–5.053) | <0.001 | 2.647 (1.911–3.673) | <0.001 | ||
| Age | 1.825 (1.333–2.5) | <0.001 | 1.624 (1.18–2.234) | 0.003 | ||
| Gender | 0.941 (0.691–1.283) | 0.7 | ||||
| Grade | G1 + G2 | 1 | G1 + G2 | 1 | ||
| G3 | 1.947 (1.339–2.832) | <0.001 | G3 | 1.222 (0.823–1.813) | 0.321 | |
| G4 | 5.235 (3.521–7.787) | <0.001 | G4 | 1.616 (1.01–2.587) | 0.045 | |
| AJCC Stage | Stage I + II | 1 | Stage I + II | 1 | ||
| Stage III | 2.51 (1.713–3.678) | <0.001 | Stage III | 1.75 (1.172–2.611) | 0.006 | |
| Stage IV | 6.192 (4.341–8.833) | <0.001 | Stage IV | 3.618 (2.403–5.448) | <0.001 | |
| EMTAB Validation Cohort | ||||||
| Univariate | Multivariate | |||||
| Factors | HR (95% CI) | p value | HR (95% CI) | p value | ||
| FAMGS Risk Score | 4.419 (1.872–10.431) | <0.001 | 2.964 (1.073–8.184) | 0.036 | ||
| Age | 2.262 (0.891–5.747) | 0.086 | 1.717 (0.604–4.823) | 0.313 | ||
| Gender | 0.441 (0.131–1.486) | 0.187 | ||||
| Grade | G1 + G2 | 1 | G1 + G2 | 1 | ||
| G3 | 3.015 (1.247–7.288) | 0.014 | G3 | 1.193 (0.425–3.347) | 0.738 | |
| G4 | 12.378 (3.222–47.557) | <0.001 | G4 | 3.277 (0.699–15.351) | 0.132 | |
| AJCC Stage | Stage I + II | 1 | Stage I + II | 1 | ||
| Stage III | 5.651 (1.985–16.081) | 0.001 | Stage III | 3.284 (1.084–9.948) | 0.036 | |
| Stage IV | 9.298 (3.551–24.341) | <0.001 | Stage IV | 6.246 (2.116–18.438) | <0.001 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhang, D.; Hu, X. A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma. Cancers 2022, 14, 4943. https://doi.org/10.3390/cancers14194943
Zhang H, Zhang D, Hu X. A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma. Cancers. 2022; 14(19):4943. https://doi.org/10.3390/cancers14194943
Chicago/Turabian StyleZhang, He, Di Zhang, and Xiaopeng Hu. 2022. "A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma" Cancers 14, no. 19: 4943. https://doi.org/10.3390/cancers14194943