
Clone Phylogenetics Reveals Metastatic Tumor Migrations, Maps, and Models
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Reconstruction of Clone Phylogenies
2.3. Inference of Tumor Migration Histories
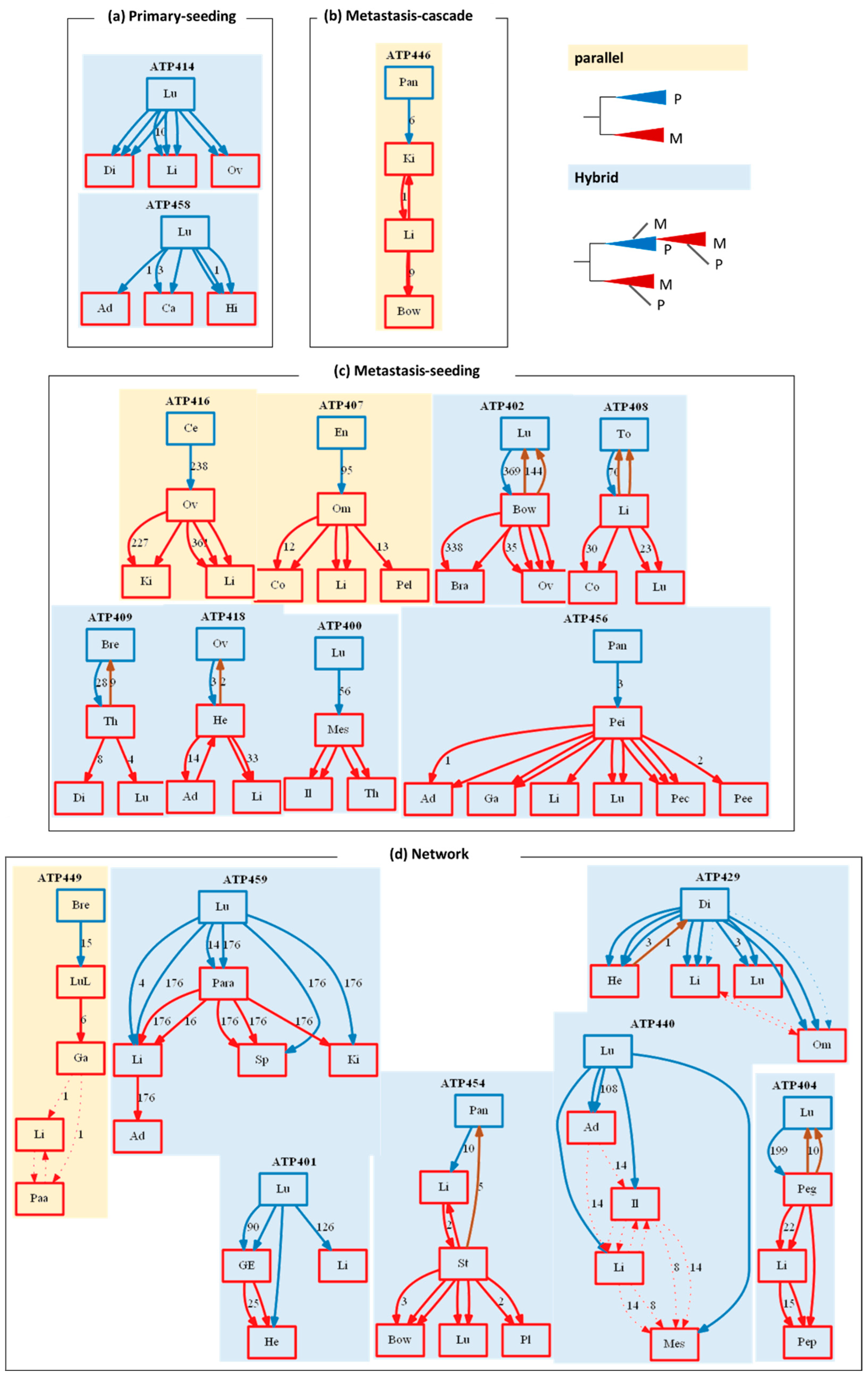
3. Results and Discussion
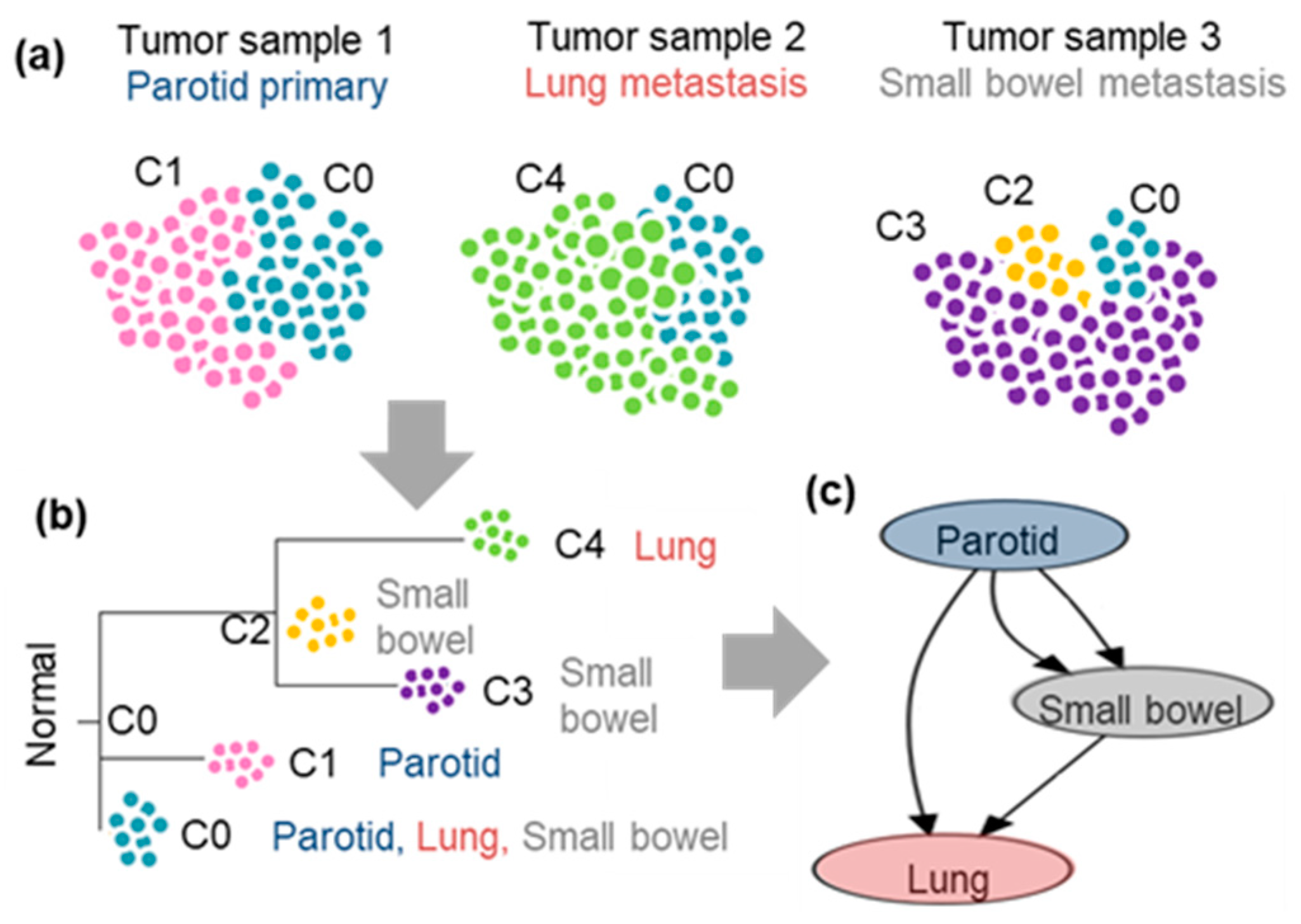
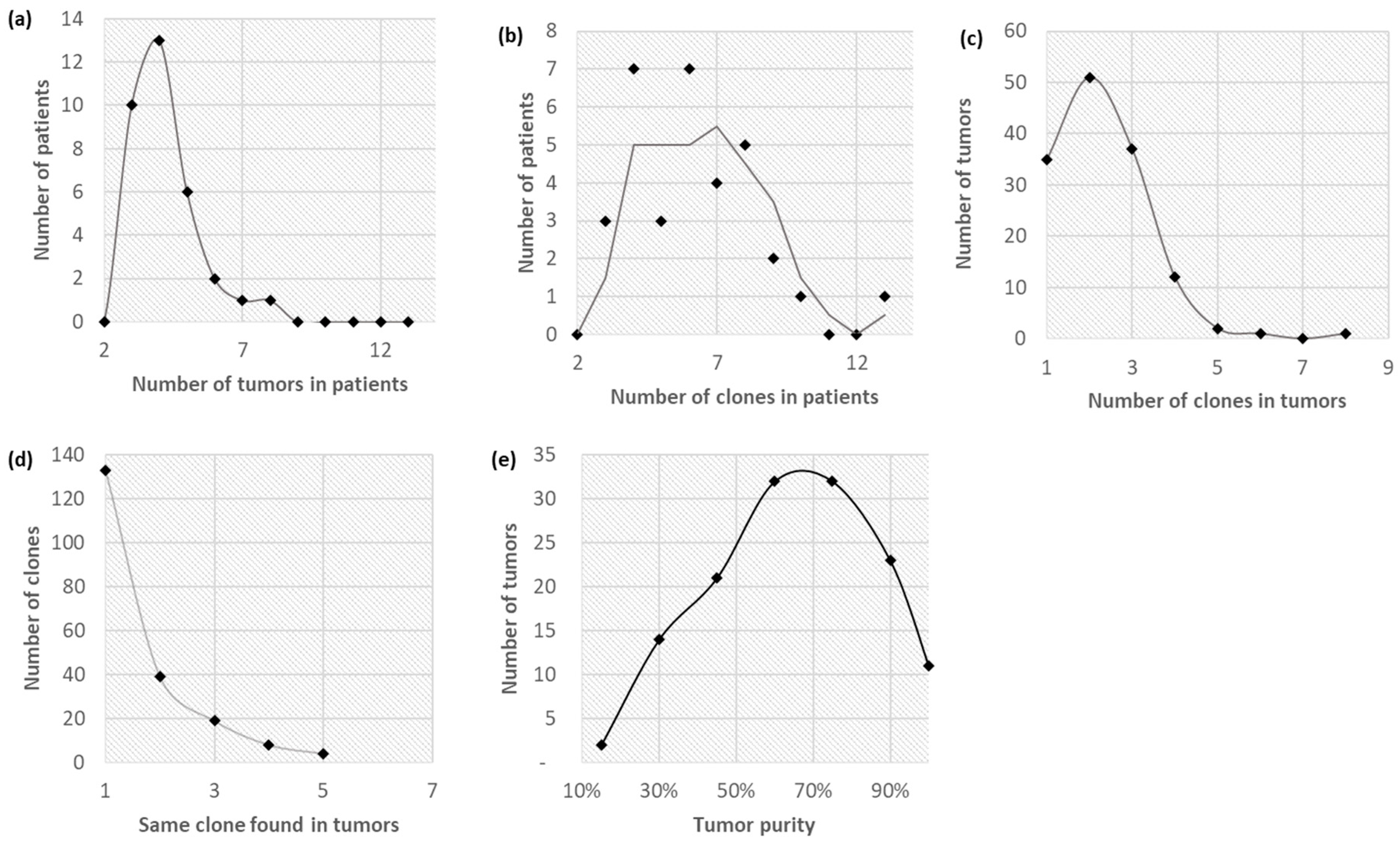
3.1. Clonal Composition of Tumors
3.2. Clone Phylogenies
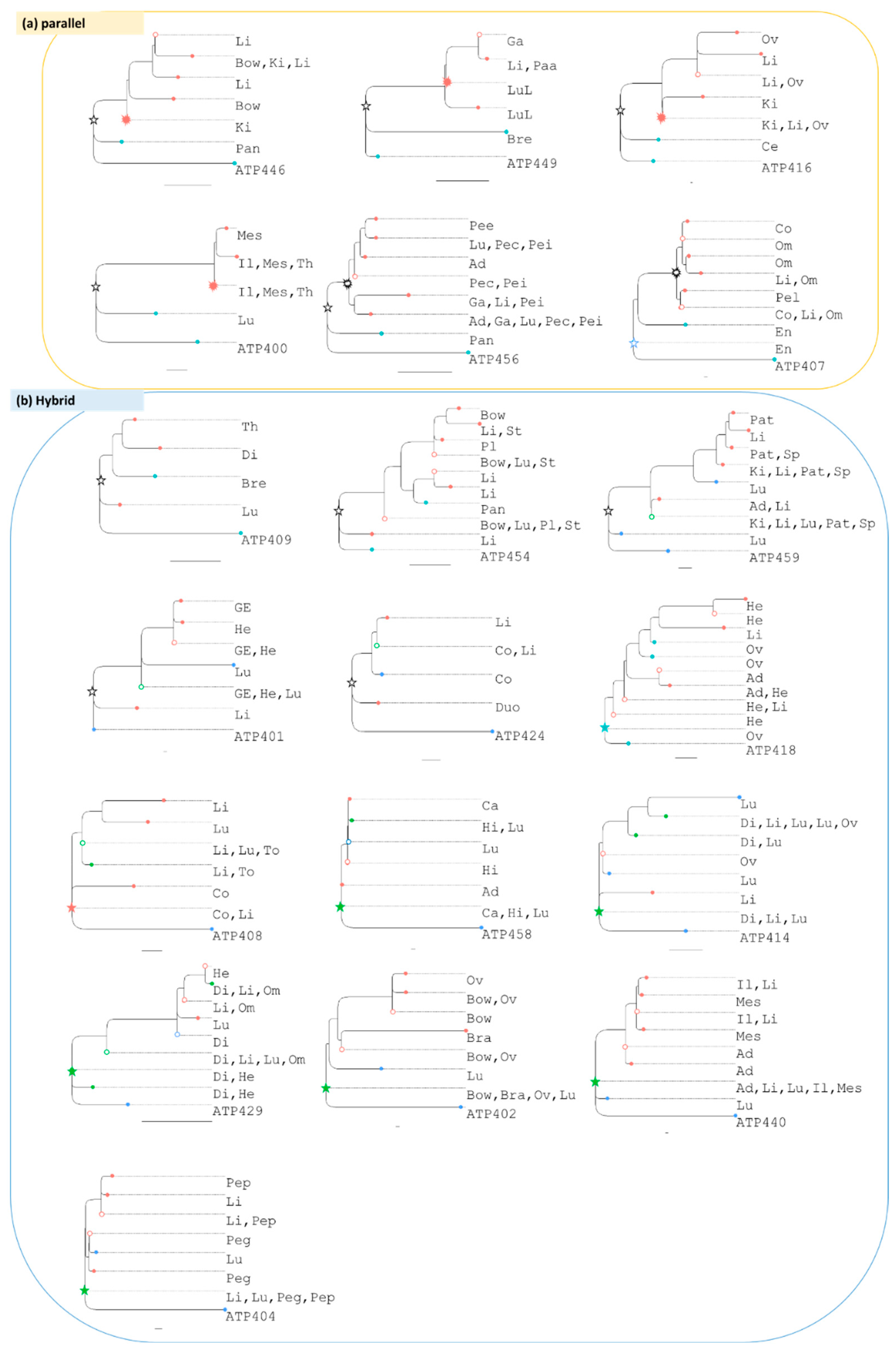
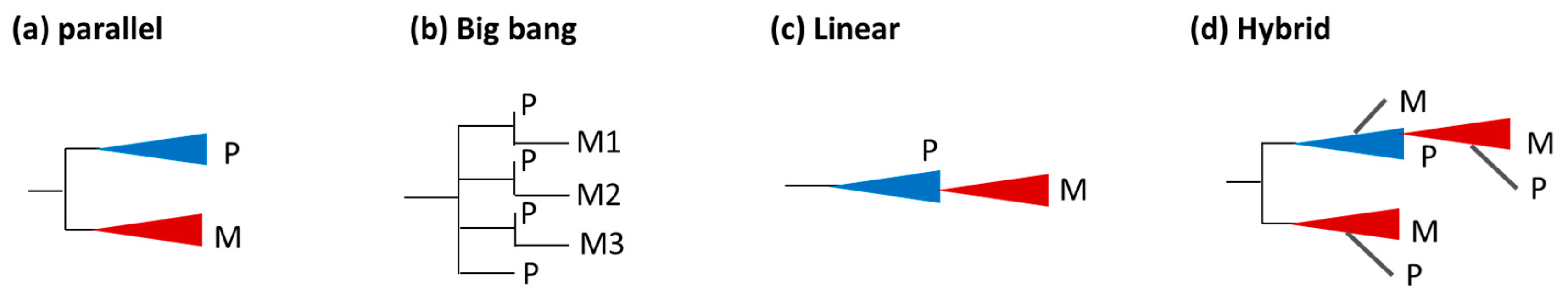
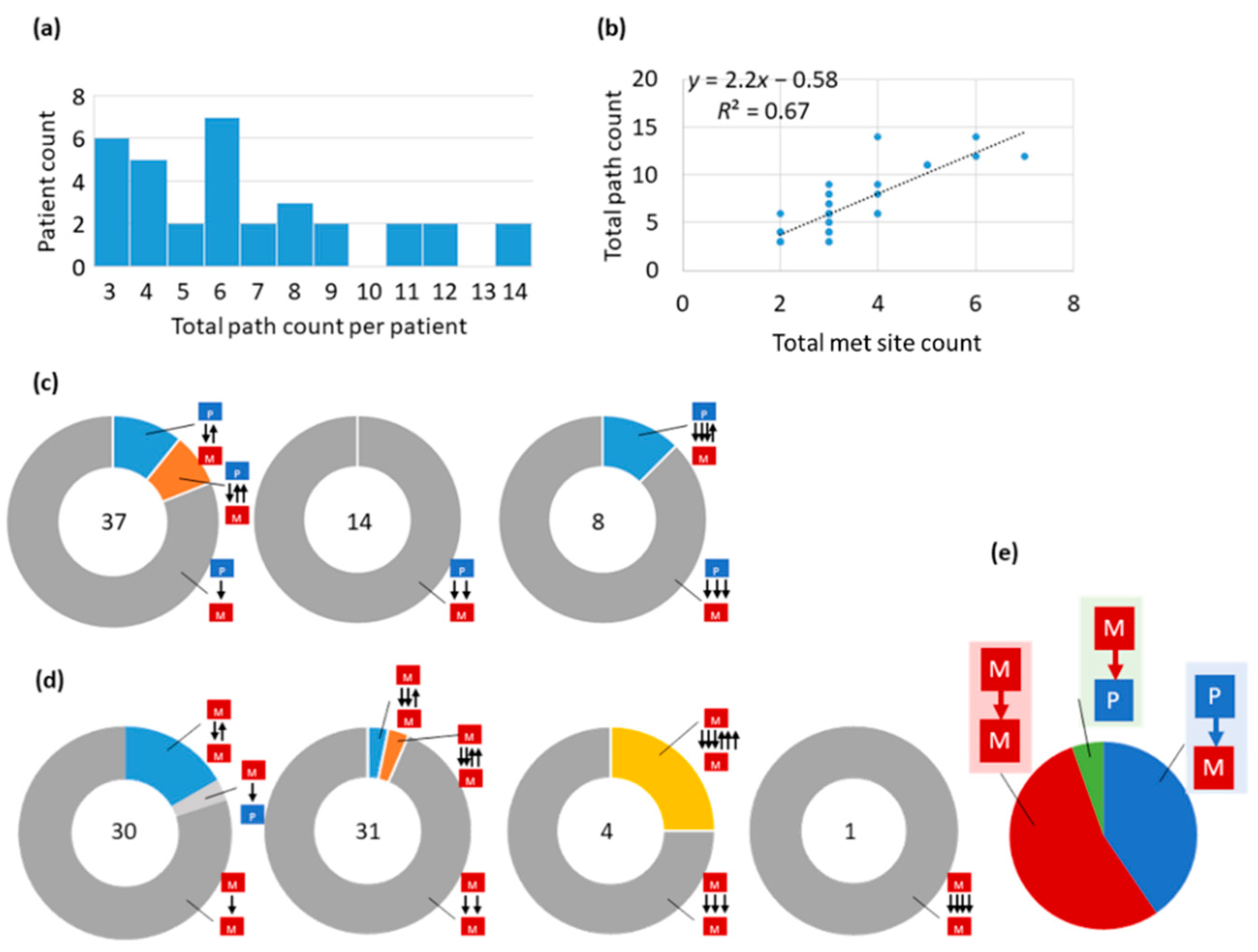
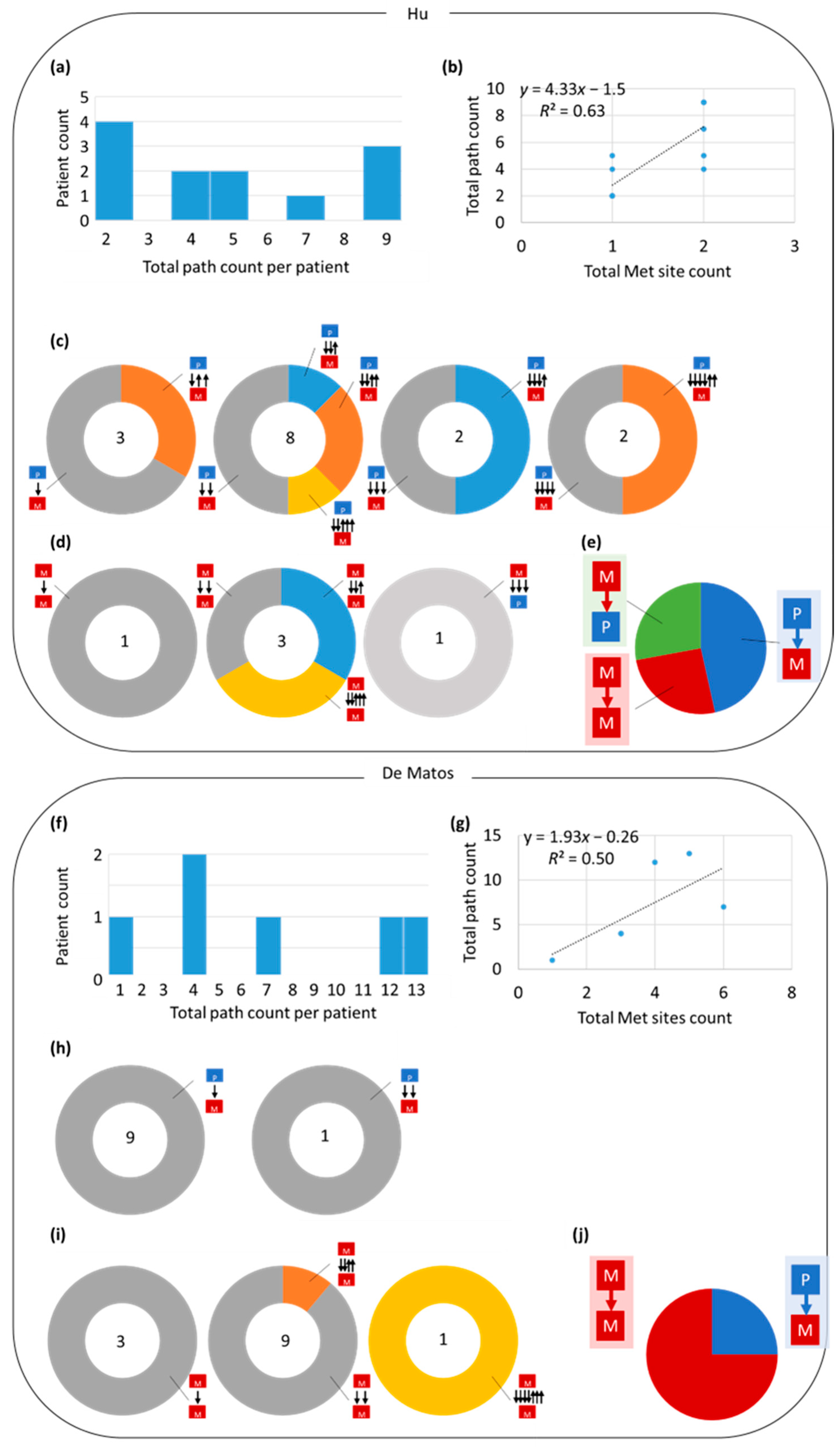
3.3. Tumor Migration Histories
3.4. Preponderance of Each Type of Clonal Migration and Exchange
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Noorani, A.; Li, X.; Goddard, M.; Crawte, J.; Alexandrov, L.B.; Secrier, M.; Eldridge, M.D.; Bower, L.; Weaver, J.; Lao-Sirieix, P.; et al. Genomic evidence supports a clonal diaspora model for metastases of esophageal adenocarcinoma. Nat. Genet. 2020, 52, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, G.; Van Loo, P.; Corcoran, N.M.; Wedge, D.C.; Markowetz, F.; Hovens, C.M. How subclonal modeling is changing the metastatic paradigm. Clin. Cancer Res. 2017, 23, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Swanton, C. Metastasis as an evolutionary process. Science 2016, 352, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.M.; Zhao, B.; Bai, Y.; Iamarino, A.; Gaffney, S.G.; Schlessinger, J.; Lifton, R.P.; Rimm, D.L.; Townsend, J.P. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Fisk, J.N.; Mahal, A.R.; Dornburg, A.; Gaffney, S.G.; Aneja, S.; Contessa, J.N.; Rimm, D.; Yu, J.B.; Townsend, J.P. Premetastatic shifts of endogenous and exogenous mutational processes support consolidative therapy in EGFR-driven lung adenocarcinoma. Cancer Lett. 2022, 526, 346–351. [Google Scholar] [CrossRef]
- Hong, W.S.; Shpak, M.; Townsend, J.P. Inferring the origin of metastases from cancer phylogenies. Cancer Res. 2015, 75, 4021–4025. [Google Scholar] [CrossRef]
- Kumar, S.; Chroni, A.; Tamura, K.; Sanderford, M.; Oladeinde, O.; Aly, V.; Vu, T.; Miura, S. PathFinder: Bayesian inference of clone migration histories in cancer. Bioinformatics 2020, 36, i675–i683. [Google Scholar] [CrossRef]
- El-Kebir, M.; Satas, G.; Raphael, B.J. Inferring parsimonious migration histories for metastatic cancers. Nat. Genet. 2018, 50, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Sottoriva, A.; Graham, T.A. Measuring Clonal Evolution in Cancer with Genomics. Annu. Rev. Genom. Hum. Genet. 2019, 20, 309–329. [Google Scholar] [CrossRef]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated multiregional analysis proposing a new model of colorectal cancer evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef] [PubMed]
- Somarelli, J.A.; Gardner, H.; Cannataro, V.L.; Gunady, E.F.; Boddy, A.M.; Johnson, N.A.; Fisk, J.N.; Gaffney, S.G.; Chuang, J.H.; Li, S.; et al. Molecular Biology and Evolution of Cancer: From Discovery to Action. Mol. Biol. Evol. 2020, 37, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Chroni, A.; Vu, T.; Miura, S.; Kumar, S. Delineation of Tumor Migration Paths by Using a Bayesian Biogeographic Approach. Cancers 2019, 11, 1880. [Google Scholar] [CrossRef]
- Chroni, A.; Miura, S.; Oladeinde, O.; Aly, V.; Kumar, S. Migrations of cancer cells through the lens of phylogenetic biogeography. Sci. Rep. 2021, 11, 17184. [Google Scholar] [CrossRef]
- Alves, J.M.; Prado-Lopez, S.; Cameselle-Teijeiro, J.M.; Posada, D. Rapid evolution and biogeographic spread in a colorectal cancer. Nat. Commun. 2019, 10, 5139. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Gomez, K.; Murillo, O.; Huuki, L.A.; Vu, T.; Buturla, T.; Kumar, S. Predicting clone genotypes from tumor bulk sequencing of multiple samples. Bioinformatics 2018, 34, 4017–4026. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Sammut, S.J.; Ross, E.M.; Bashford-Rogers, R.; Greenstein, E.; Markus, H.; Morganella, S.; Teng, Y.; Maruvka, Y.; Pereira, B.; et al. The Genomic and Immune Landscapes of Lethal Metastatic Breast Cancer. Cell Rep. 2019, 27, 2690–2708. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594. [Google Scholar] [CrossRef]
- Wei, Q.; Ye, Z.; Zhong, X.; Li, L.; Wang, C.; Myers, R.E.; Palazzo, J.P.; Fortuna, D.; Yan, A.; Waldman, S.A.; et al. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann. Oncol. 2017, 28, 2135–2141. [Google Scholar] [CrossRef]
- Chen, H.N.; Shu, Y.; Liao, F.; Liao, X.; Zhang, H.; Qin, Y.; Wang, Z.; Luo, M.; Liu, Q.; Xue, Z.; et al. Genomic evolution and diverse models of systemic metastases in colorectal cancer. Gut 2022, 71, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Cresswell, G.D.; Nichol, D.; Spiteri, I.; Tari, H.; Zapata, L.; Heide, T.; Maley, C.C.; Magnani, L.; Schiavon, G.; Ashworth, A.; et al. Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking. Nat. Commun. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Gui, P.; Bivona, T.G. Evolution of metastasis: New tools and insights. Trends Cancer 2022, 8, 98–109. [Google Scholar] [CrossRef]
- Ryser, M.D.; Mallo, D.; Hall, A.; Hardman, T.; King, L.M.; Tatishchev, S.; Sorribes, I.C.; Maley, C.C.; Marks, J.R.; Hwang, E.S.; et al. Minimal barriers to invasion during human colorectal tumor growth. Nat. Commun. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Vu, T.; Deng, J.; Buturla, T.; Oladeinde, O.; Choi, J.; Kumar, S. Power and pitfalls of computational methods for inferring clone phylogenies and mutation orders from bulk sequencing data. Sci. Rep. 2020, 10, 3498. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing Core of Molecular Evolutionary Genetics Analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chroni, A.; Miura, S.; Hamilton, L.; Vu, T.; Gaffney, S.G.; Aly, V.; Karim, S.; Sanderford, M.; Townsend, J.P.; Kumar, S. Clone Phylogenetics Reveals Metastatic Tumor Migrations, Maps, and Models. Cancers 2022, 14, 4326. https://doi.org/10.3390/cancers14174326
Chroni A, Miura S, Hamilton L, Vu T, Gaffney SG, Aly V, Karim S, Sanderford M, Townsend JP, Kumar S. Clone Phylogenetics Reveals Metastatic Tumor Migrations, Maps, and Models. Cancers. 2022; 14(17):4326. https://doi.org/10.3390/cancers14174326
Chicago/Turabian StyleChroni, Antonia, Sayaka Miura, Lauren Hamilton, Tracy Vu, Stephen G. Gaffney, Vivian Aly, Sajjad Karim, Maxwell Sanderford, Jeffrey P. Townsend, and Sudhir Kumar. 2022. "Clone Phylogenetics Reveals Metastatic Tumor Migrations, Maps, and Models" Cancers 14, no. 17: 4326. https://doi.org/10.3390/cancers14174326