1. Introduction
Liver cancer is one of the most frequent, and the fourth most lethal, cancer around the globe [
1]. Hepatocellular carcinoma (HCC) is the most common primary hepatic malignant tumor and accounts for more than 80% of all liver cancers worldwide. Despite the advances in therapy, patients with HCC still have poor outcome, especially those at advanced stages [
2]. Therefore, early diagnosis and risk stratification of prognosis are of great significance for patients with liver cancer, and initiating effective treatment expeditiously may efficiently improve their prognosis [
3]. Unfortunately, real-world utilization of biomarkers to predict clinical outcome of HCC patients by precise classification and specific clinical decision-making remains urgent but still lacking.
In the past two decades, the relevant research communities have made great strides in microarray, high-throughput, and multi-omics technologies. One of the most widely adopted applications of these technologies is to generate a large number of cancer gene expression-associated signatures, including HCC gene signatures [
4,
5]. Plenty of HCC prognostic gene expression signatures have been described. As one of the pioneering examples, Lee et al. analyzed gene expression profiling data of 91 HCC patients and identified two distinctive subclasses that are highly associated with the survival of patients [
6]. Subsequently multiple signatures were reported to be associated with survival [
7,
8], recurrence [
9,
10], metastasis [
11], and other clinical parameters.
However, even though HCC gene signatures have been studied extensively, none of the published HCC-dependent genetic signatures have entered clinical practice. This is an issue that also remains challenging in other cancer entities, e.g., breast cancer (BC) and colorectal cancer (CRC) [
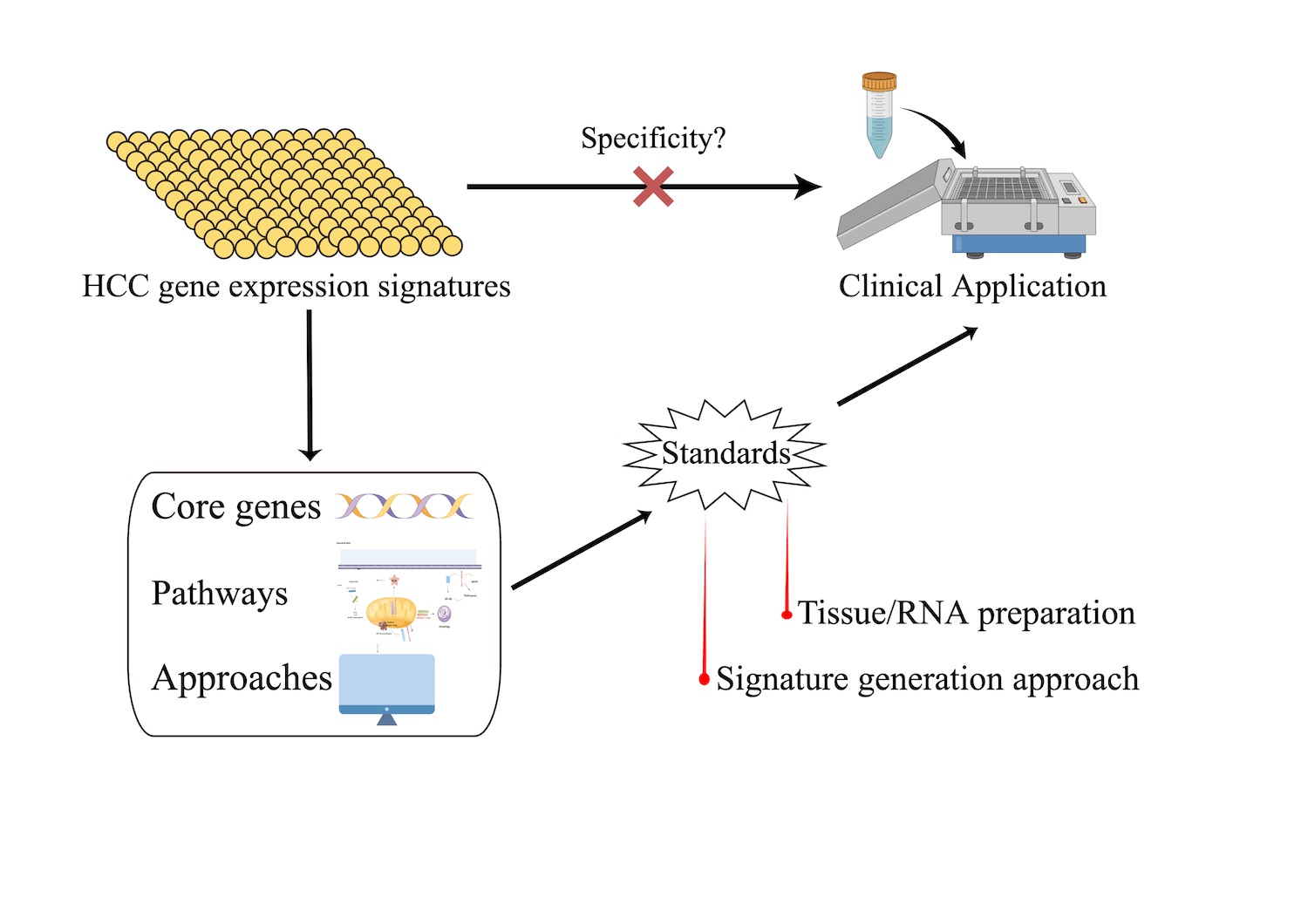
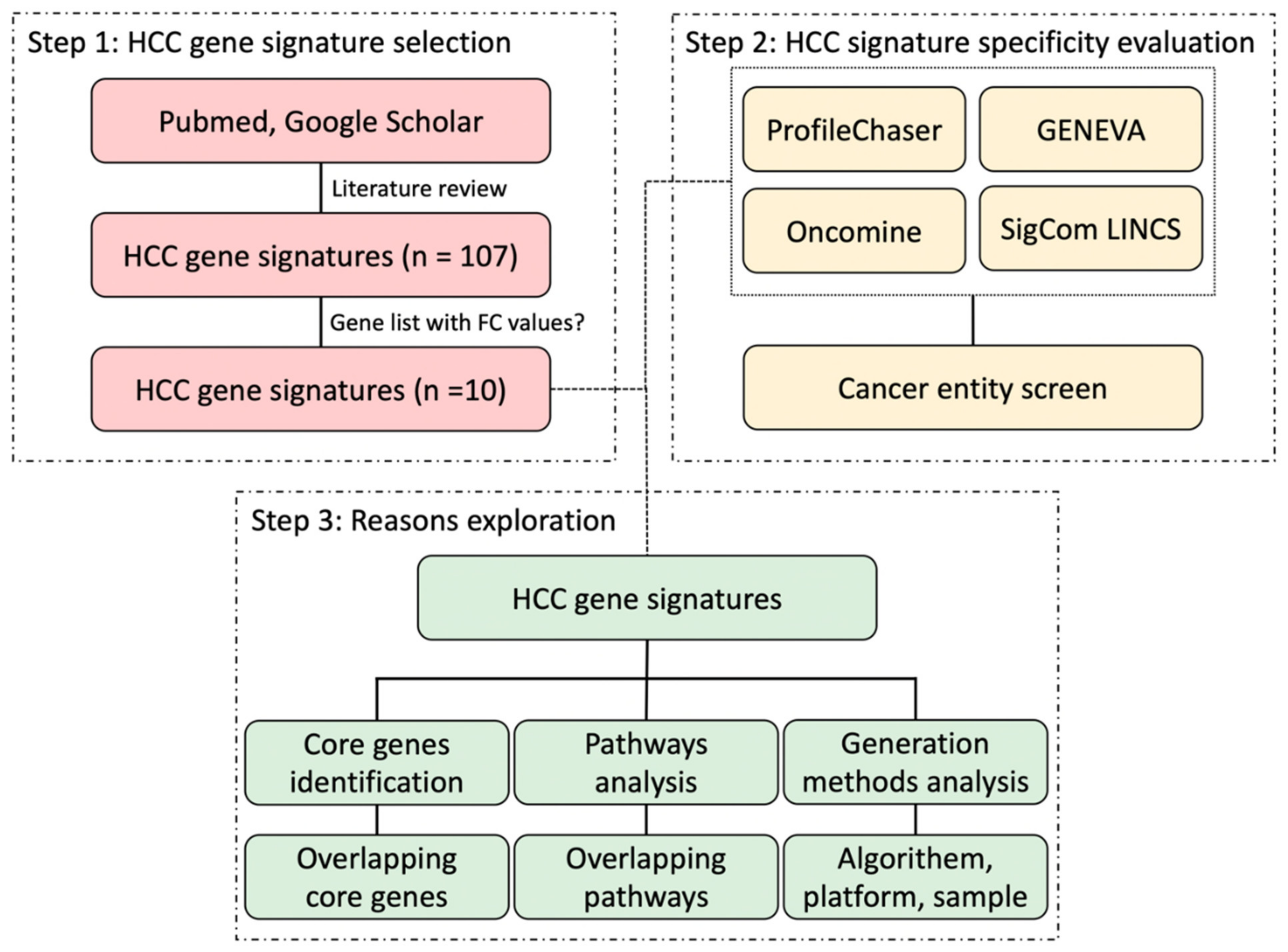
5]. Although many authors addressed the robustness and sensitivity of their molecular-based HCC biomarkers when identifying these gene signatures, they mostly did not widely evaluate the specificity of HCC gene signatures, which is also a crucial aspect of applying HCC gene signatures into clinical routines. In this study, mainly by using four public and commercial gene expression comparison tools, we aimed to explore the specificity of HCC gene signatures, compared it with gene signatures of other cancer entities, and aimed to investigate the difficulties associated with these approaches.
4. Discussion
The profound heterogeneity of HCC is becoming increasingly clear. The intra-tumor heterogeneity of HCC leads to the lack of robustness and reproducibility of molecular biomarkers [
32], and meanwhile random gene sets show prognostic power for patients with HCC [
33]. This may also be one of the major obstacles in establishing valid gene expression signatures for the diagnosis, prognosis, and response to treatment in patients with HCC.
Gene expression profiling creates a panoramic view of cellular function by measuring the expression of thousands of genes at once, which makes it feasible to detect and classify genetic changes in cells in the form of gene expression signatures [
5]. Although the advancements have been made, successful translation of gene expression profiling into clinical applications remains challenging. The limitations include not only the need for further optimization of tissue preparation and storage procedures, but also the need for sufficient bioinformatics strategies and standardized independent validation of gene expression signatures [
4].
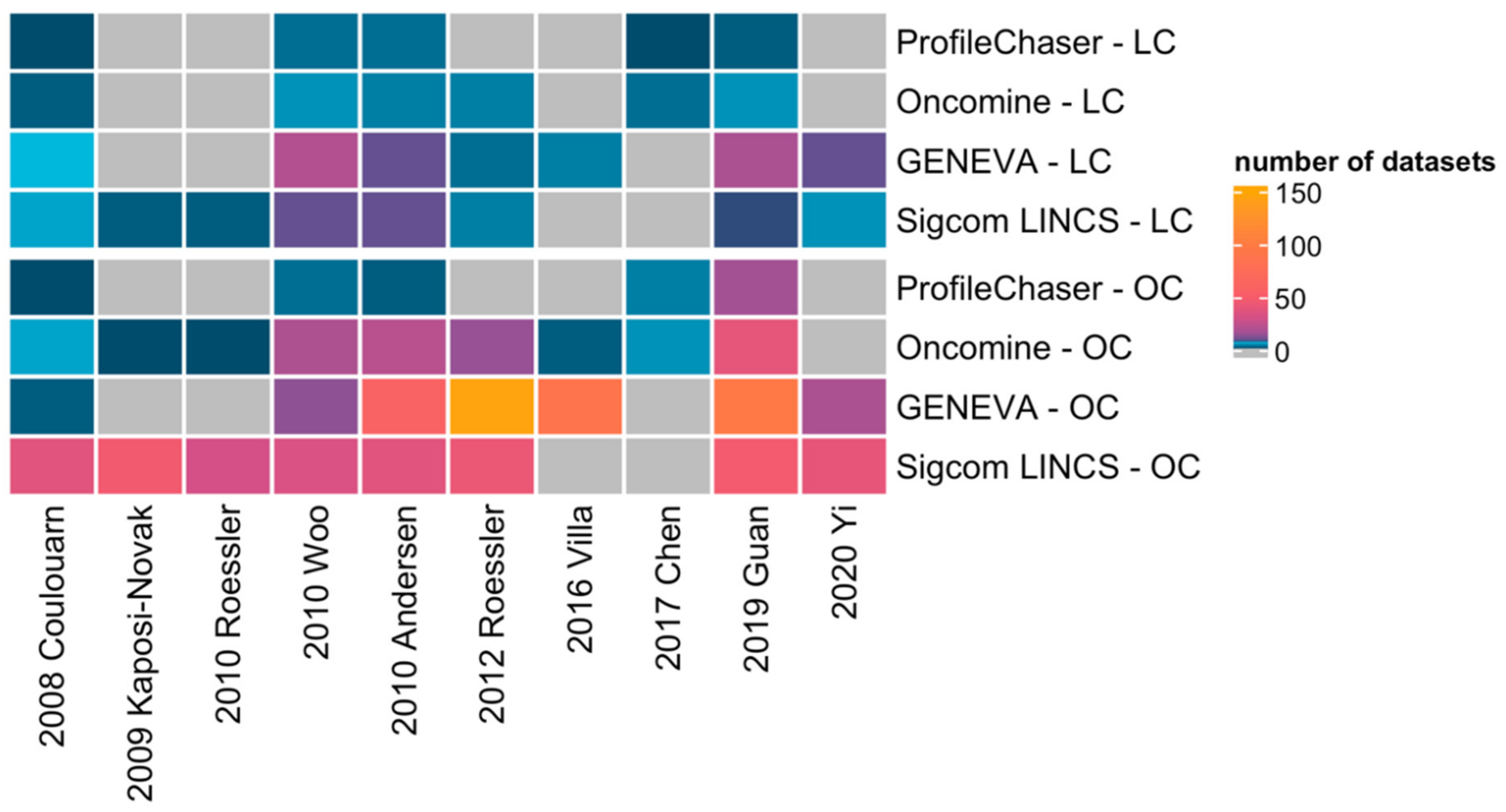
However, up to now no global comparison of the available gene expression signatures has been attempted. Therefore, by means of implementing four comparison bio-tools, we successfully validated in this study that current HCC gene signatures are unspecific for HCC, which may have several underlying reasons.
In this study, the results of similar expression profiles search vary among the four profiling tools. On one hand, how the four tools perform gene signature query is quite different: ProfileChaser compares gene expression profiles by weighted correlation coefficient, Oncomine performs association analysis, GENEVA identifies the variance of gene expression, and Sigcom LINCS does signature enrichment analysis. On the other hand, the composition of databases on which the four tools are based is also different: while datasets in ProfileChaser and Oncomine are composed of microarray data, GENEVA and Sigcom LINCS perform signature search based on RNA-seq data.
Unlike HCC gene signatures, some BC and CRC gene signatures are already commercially or clinically available. Our study shows that not only HCC gene signatures, but the majority of these available BC and CRC gene signatures (5/7) also got many different cancer matches after running in Oncomine. This suggests that even though these gene signatures have been commercialized or entered the clinic, they are still not specific enough. In fact, currently available gene signatures for early and intermediate stages of CRC should only be used in specific clinical settings due to the lack of a plausible biological interpretability and have no predictive value of treatment benefit [
5,
34]. Although the promising CRC classifier based on gene expression, the consensus molecular subtypes (CMS) classification system, shows prognostic value in intermediate and advanced-stage CRC, there is still a lack of standardization and a requirement for bioinformatics resources [
5]. BC has a leading edge in the clinical application of gene signatures, but there is still debate about their use in BC, especially in early BC with positive lymph nodes [
5]. In addition, Manjang and co-workers also demonstrated that the prognostic BC gene signatures lack a clear biological meaning [
35], suggesting us to notice the divergences at the level of gene patterns and gene expression profiles.
In order to overcome those obstacles, recent studies revealed that long noncoding RNAs (lncRNAs), due to abnormalities in chromatin modification and alternative splicing, play an important role in tumor development and progression and are more likely cancer-type specific compared with protein-coding genes [
36,
37,
38]. The gene signatures included in our study were all protein-coding genes, as these were commonly utilized in gene expression profiling in the past and available in respective databases and evaluation tools. However, further exploration of lncRNA signatures may provide additional insights into molecular mechanism, ultimately leading to progress in the management of HCC.
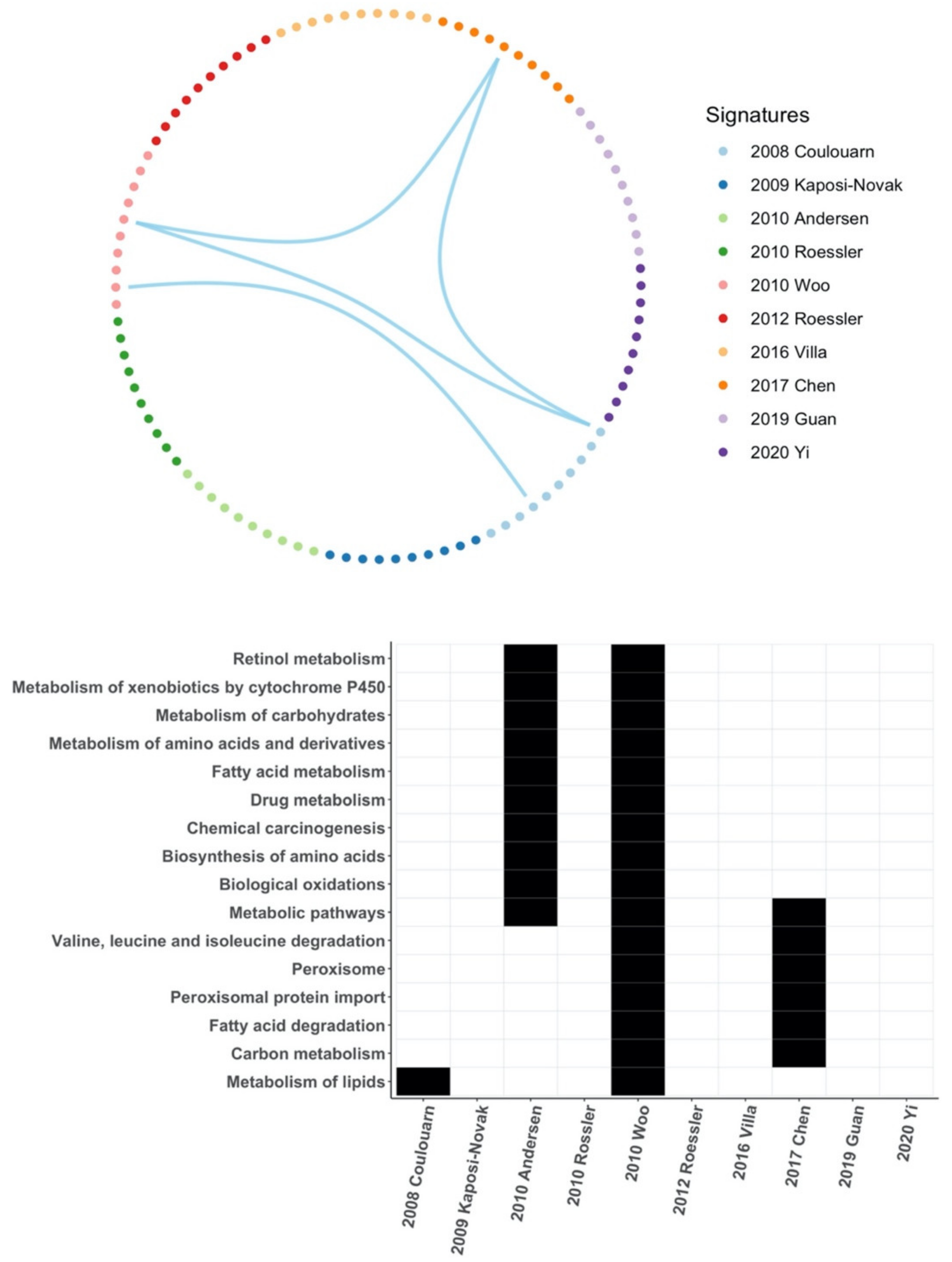
Core genes or hub genes are at the core of the regulatory network and play an important role in the biological classification of samples by gene signatures. In this study, all core genes were unique to each HCC gene signature and only a few signal pathways overlapped between few HCC gene signatures, which indicates that they may produce very different biological classifications. This accounts for the differences between those HCC gene signatures.
Benefiting from the bioinformatic technology advances, the scientific community has developed a vast number of methods to generate gene signatures. Different platforms, algorithms, and sources of samples play an important role in the generation of cancer gene signatures. In our study, taking the above three factors into consideration, all the selected HCC gene signatures have different generation methods. Meanwhile, these technical differences and variance in samples have led to biological limitations of HCC gene signatures. A standardized method and procedure are needed to construct more interpretable and comparable gene signatures.
Our study demonstrates that current HCC genetic signatures are not good enough for prognosis and independent validation of HCC gene signatures remains challenging, which may be critical for HCC gene signatures to enter clinical application. Overall, due to the heterogeneity of HCC, our data suggest the necessity of standards, not only in data format and tissue storage, but also in signature generation, statistical methods, and independent validation.
Due to enabling reproducible re-analysis of functional genomics data, the scientific community has long been aware of the need to standardize the data format when describing a microarray or sequencing study. The Functional Genomics Data (FGED) Society proposed two recording and reporting standards successively: the Minimum Information About a Microarray Experiment (MIAME) [
39] and the Minimum Information about a high-throughput SEQuencing Experiment (MINSEQE) [
40]. The two guidelines both emphasized the importance of providing the following information to make the data understandable and reusable: (1) raw data and final processed data; (2) general information about the experiment; (3) sample annotation, the experimental factors, and their values; (4) laboratory and data processing protocols; and (5) sample data relationships [
40,
41]. Data repositories such as GEO and ArrayExpress both employed MIAME and MINSEQE as standards for data depositing, facilitating data submission/sharing and usage.
Sample storage is another critical factor for gene expression profiling as extraction of high-quality RNA from the samples is a prerequisite for reliable measurement results [
42,
43]. While cryopreserved cancer tissue had no adverse effect on RNA quality, RNA degradation depended on the time the samples were not frozen [
44]. Uniform standards for sample storage are not well established, which may also account for the inconsistent findings among different studies, but a few basic guidelines are recommended. First, patient samples need to be frozen and stored at −80 °C as soon as possible, preferably with cold ischemia less than 1 h [
44]. Second, RNA stabilization reagents, i.e., RNAlater, are recommended to preserve RNA integrity during frozen storage [
45]. Third, samples should be transported on ice or in liquid nitrogen to avoid significant degradation of RNA quality before freezing them. Fourth, samples can be frozen in many small sections and stored in separate partitions to reduce the effects of repeated freezing and thawing and temperature fluctuations.
A variety of gene signature generation methods exist in different studies and they are evolving more and more sophisticated, contributing to the limited reproducibility and comparability of HCC gene signatures. No standards for gene signature generation algorithm have been established, yet we would like to propose that the algorithms should meet two criteria: (1) the algorithms need to select genes that are not only statistically significant but also biologically meaningful, and (2) the algorithms should maintain their effectiveness on different sequencing platforms and samples from different sources to rule out that they are platform-specific or sample-specific. In addition, the importance of independent validation is always in need of emphasizing.







{kind=link}
{kind=link}
{kind=link}
{kind=link}