
T-Cell-Based Cellular Immunotherapy of Multiple Myeloma: Current Developments

Abstract
:Simple Summary
Abstract
1. Introduction
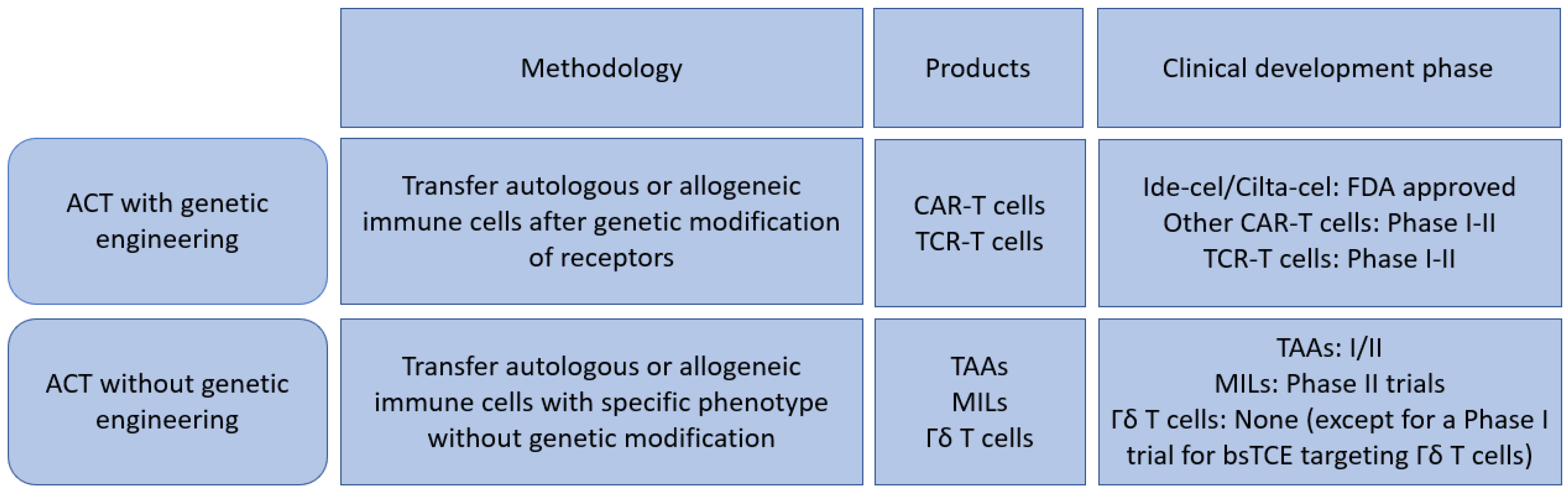
2. Genetically Engineered T Cells
2.1. Chimeric Antigen Receptor (CAR) T Cells
2.1.1. NCI
2.1.2. UPENN BCMA
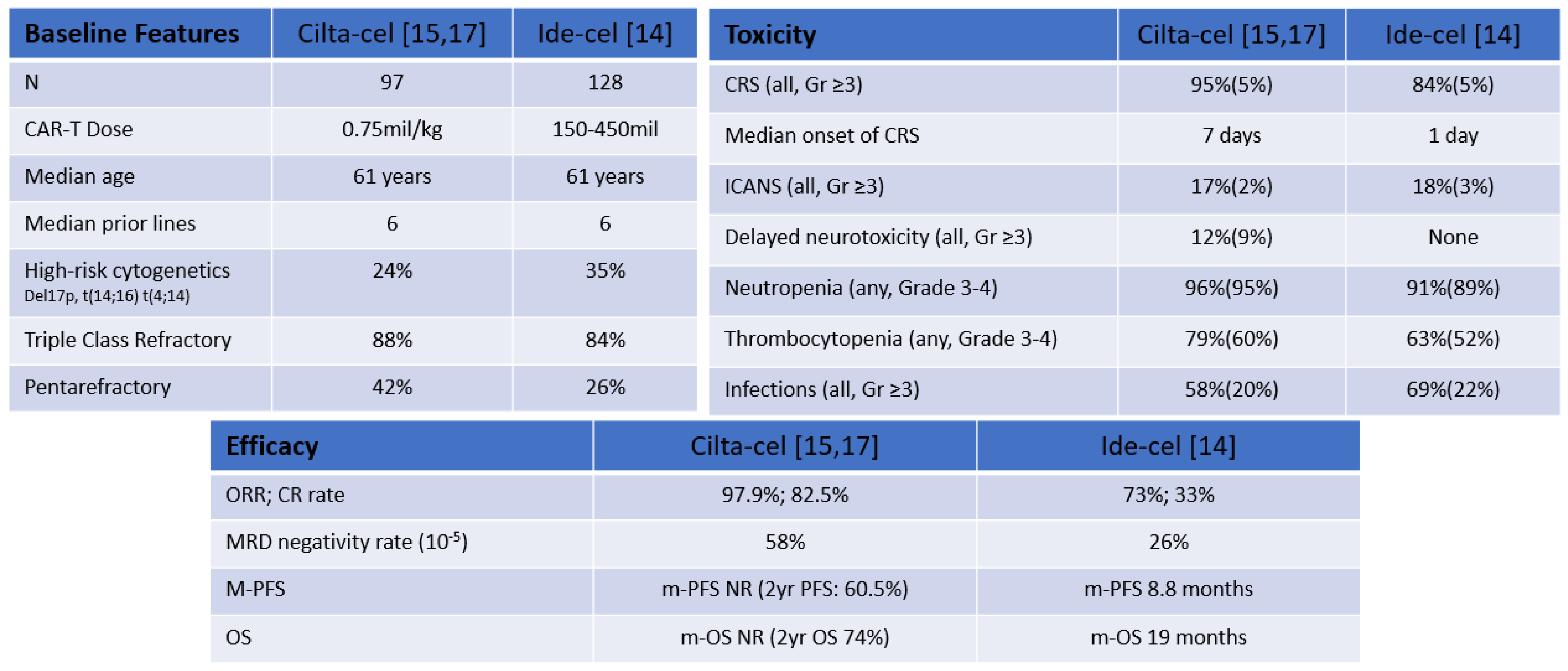
2.1.3. Idecabtagene Vicleucel (bb2121)
2.1.4. Bb21217
2.1.5. LCAR-B38 M
2.1.6. Ciltacabtagene Autoleucel
2.1.7. Orvacabtagene Autoleucel (Orva-Cel)
2.1.8. Zevorcabtagene Autoleucel (Zevor-Cel)
2.1.9. ALLO-715
2.2. Associated CAR T Cells Toxicities
2.3. TCR (T Cell Receptor) Engineered T Cells
2.3.1. NCT01352286 (SPEAR T Cells)
2.3.2. NCT03399448 (NYCE T Cells)
2.3.3. Miscellaneous
3. Non-Genetically Modified Strategies
3.1. Multi Tumor Associated Antigen Targeted T Cells (TAAs)
3.2. Marrow Infiltrating Lymphocytes (MILs)
γδ. T Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal Gammopathy of Undetermined Significance (MGUS) and Smoldering Multiple Myeloma (SMM): Novel Biological Insights and Development of Early Treatment Strategies. Blood 2011, 117, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Kyle, R.A.; Rajkumar, S.V. From Myeloma Precursor Disease to Multiple Myeloma: New Diagnostic Concepts and Opportunities for Early Intervention. Clin. Cancer Res. 2011, 17, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Mateos, M.V.; Sanchez-Abarca, L.I.; Puig, N.; Vidriales, M.B.; López-Corral, L.; Corchete, L.A.; Hernandez, M.T.; Bargay, J.; De Arriba, F.; et al. Immune Status of High-Risk Smoldering Multiple Myeloma Patients and Its Therapeutic Modulation under Lendex: A Longitudinal Analysis. Blood 2016, 127, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Leblay, N.; Maity, R.; Hasan, F.; Neri, P. Deregulation of Adaptive T Cell Immunity in Multiple Myeloma: Insights Into Mechanisms and Therapeutic Opportunities. Front. Oncol. 2020, 10, 636. [Google Scholar] [CrossRef]
- Romano, A.; Conticello, C.; Cavalli, M.; Vetro, C.; La Fauci, A.; Parrinello, N.L.; Di Raimondo, F. Immunological Dysregulation in Multiple Myeloma Microenvironment. Biomed. Res. Int. 2014, 2014, 198539. [Google Scholar] [CrossRef]
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in Overall Survival and Costs of Multiple Myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022, 28, 1759–1764. [Google Scholar] [CrossRef]
- Pittari, G.; Vago, L.; Festuccia, M.; Bonini, C.; Mudawi, D.; Giaccone, L.; Bruno, B. Restoring Natural Killer Cell Immunity against Multiple Myeloma in the Era of New Drugs. Front. Immunol. 2017, 8, 1444. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT Immune Checkpoint Blockade Restores CD81 T-Cell Immunity against Multiple Myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Mikkilineni, L.; Kochenderfer, J.N. CAR T Cell Therapies for Patients with Multiple Myeloma. Nat. Rev. Clin. Oncol. 2021, 18, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Rationale for Targeting and Current Therapeutic Approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; et al. Updated Results from CARTITUDE-1: Phase 1b/2Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Patients With Relapsed/Refractory Multiple Myeloma. Blood 2021, 138, 549. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-Cell Maturation Antigen Is a Promising Target for Adoptive T-Cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T Cells Expressing an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B Cell Maturation Antigen-Specific CAR T Cells Are Clinically Active in Multiple Myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of Cytokine Release Syndrome Associated with the CAR T Cell Therapy Tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen–Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Lin, Y.; Raje, N.S.; Berdeja, J.G.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Massaro, M.; et al. Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy, in Patients with Relapsed and Refractory Multiple Myeloma: Updated Results from Phase 1 CRB-401 Study. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Shah, N.; Munshi, N.C.; Berdeja, J.G.; Jagannath, S.; Finney, O.; Martin, N.; Agarwal, A.; Rowe, E.; Campbell, T.B.; San-Miguel, J.F. Baseline Correlates of Complete Response to Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy in Patients with Relapsed and Refractory Multiple Myeloma: Subanalysis of the KarMMa Trial. Blood 2021, 138, 1739. [Google Scholar] [CrossRef]
- Anderson, L.D., Jr.; Munshi, N.C.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.D.; Lin, Y.; Oriol, A.; et al. Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy, in Relapsed and Refractory Multiple Myeloma: Updated KarMMa Results. J. Clin. Oncol. 2021, 39, 8016. [Google Scholar] [CrossRef]
- Delforge, M.; Baz, R.C.; Cavo, M.; Callander, N.S.; Ghobadi, A.; Rodriguez-Otero, P.; Mateos, M.-V.; Massaro, M.; Ding, L.; Patel, P.; et al. KarMMa-3: A Phase 3 Study of Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy Vs Standard Regimens in Relapsed and Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Berdeja, J.G.; Truppel-Hartmann, A.; Casadebaig, M.-L.; Wortman-Vayn, H.; Shelat, S.G.; Novick, S.; Shah, N. KarMMa-4: Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T-Cell Therapy, in High-Risk Newly Diagnosed Multiple Myeloma. Blood 2020, 136, 18–19. [Google Scholar] [CrossRef]
- Raje, N.S.; Berdeja, J.G.; Rodriguez-Otero, P.; Green, D.J.; Jagannath, S.; Lonial, S.; Gipson, A.; Caia, A.; Martin, N.; Yang, Z.; et al. KarMMa-7, a Phase 1/2, Dose-Finding and Dose-Expansion Study of Combination Therapies with Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy for Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2021, 138, 4830. [Google Scholar] [CrossRef]
- Raje, N.S.; Shah, N.; Jagannath, S.; Kaufman, J.L.; Siegel, D.S.; Munshi, N.C.; Rosenblatt, J.; Lin, Y.; Jakubowiak, A.; Timm, A.; et al. Updated Clinical and Correlative Results from the Phase I CRB-402 Study of the BCMA-Targeted CAR T Cell Therapy Bb21217 in Patients with Relapsed and Refractory Multiple Myeloma. Blood 2021, 138, 548. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A Phase 1, Open-Label Study of LCAR-B38M, a Chimeric Antigen Receptor T Cell Therapy Directed against B Cell Maturation Antigen, in Patients with Relapsed or Refractory Multiple Myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, L.-J.; Yang, S.-S.; Sun, Y.; Wu, W.; Liu, Y.-F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.-Q.; et al. Exploratory Trial of a Biepitopic CAR T-Targeting B Cell Maturation Antigen in Relapsed/Refractory Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-Y.; Zhao, W.-H.; Liu, J.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. Long-Term Follow-up of a Phase 1, First-in-Human Open-Label Study of LCAR-B38M, a Structurally Differentiated Chimeric Antigen Receptor T (CAR-T) Cell Therapy Targeting B-Cell Maturation Antigen (BCMA), in Patients (Pts) with Relapsed/Refractory Multiple. Blood 2019, 134, 579. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Madduri, D.; Usmani, S.Z.; Jagannath, S.; Singh, I.; Zudaire, E.; Yeh, T.-M.; Allred, A.J.; Banerjee, A.; Goldberg, J.D.; Schecter, J.M.; et al. Results from CARTITUDE-1: A Phase 1b/2 Study of JNJ-4528, a CAR-T Cell Therapy Directed Against B-Cell Maturation Antigen (BCMA), in Patients with Relapsed and/or Refractory Multiple Myeloma (R/R MM). Blood 2019, 134, 577. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Cohen, A.D.; Delforge, M.; Hillengass, J.; Goldschmidt, H.; Weisel, K.; Raab, M.-S.; Scheid, C.; Schecter, J.M.; De Braganca, K.C.; et al. Efficacy and Safety of Ciltacabtagene Autoleucel (Cilta-Cel), a B-Cell Maturation Antigen (BCMA)-Directed Chimeric Antigen Receptor (CAR) T-Cell Therapy, in Lenalidomide-Refractory Patients with Progressive Multiple Myeloma after 1-3 Prior Lines of Therap. Blood 2021, 138, 3866. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Delforge, M.; Agha, M.; Cohen, A.D.; Cohen, Y.C.; Hillengass, J.; Anguille, S.; Kerre, T.; Roeloffzen, W.; Schecter, J.M.; et al. CARTITUDE-2: Efficacy and Safety of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen (BCMA)-Directed Chimeric Antigen Receptor T-Cell Therapy, in Patients with Multiple Myeloma and Early Relapse after Initial Therapy. Blood 2021, 138, 2910. [Google Scholar] [CrossRef]
- Mailankody, S.; Htut, M.; Lee, K.P.; Bensinger, W.; Devries, T.; Piasecki, J.; Ziyad, S.; Blake, M.; Byon, J.; Jakubowiak, A. JCARH125, Anti-BCMA CAR T-Cell Therapy for Relapsed/Refractory Multiple Myeloma: Initial Proof of Concept Results from a Phase 1/2 Multicenter Study (EVOLVE). Blood 2018, 132, 957. [Google Scholar] [CrossRef]
- Colonna, L.; Navarro, G.; Devries, T.; Beckett, V.; Amsberry, A.; Radhakrishnan, A.; Piasecki, J.; Heipel, M.; Li, Y.; Kavita, U.; et al. Orvacabtagene Autoleucel (Orva-Cel; JCARH125): A Fully Human BCMA-Targeted Second-Generation CAR T Cell Product Characterized By a Predominant Central Memory Phenotype with High in Vitro and In Vivo Proliferative Potential and Sustained In Vivo Persistenc. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Piasecki, J.; Devries, T.; Radhakrishnan, A.; Li, Y.; Heipel, M.; Fox, B.A.; Beckett, V.; Cota Stirner, M.; Conte, K.; Doerr, T.; et al. Association of Baseline and Postinfusion Biomarkers with Safety and Efficacy Endpoints in Patients Treated with Orvacabtagene Autoleucel (Orva-Cel; JCARH125) in the Phase 1/2 Evolve Study (NCT03430011). Blood 2020, 136, 2–3. [Google Scholar] [CrossRef]
- Jie, J.; Hao, S.; Jiang, S.; Li, Z.; Yang, M.; Zhang, W.; Yu, K.; Xiao, J.; Meng, H.; Ma, L.; et al. Phase 1 Trial of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells in Relapsed/Refractory Multiple Myeloma. Blood 2019, 134, 4435. [Google Scholar] [CrossRef]
- Hao, S.; Jin, J.; Jiang, S.; Li, Z.; Zhang, W.; Yang, M.; Yu, K.; Wang, W.; Chen, L.; Meng, H.; et al. Two-Year Follow-up of Investigator-Initiated Phase 1 Trials of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells (CT053) in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 27–28. [Google Scholar] [CrossRef]
- Chen, W.; Fu, C.; Cai, Z.; Li, Z.; Wang, H.; Yan, L.; Wu, Y.; Shi, X.; Gao, W.; Yan, S.; et al. Sustainable Efficacy and Safety Results from Lummicar Study 1: A Phase 1/2 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Chinese Subjects with Relapsed and/or Refractory Multiple Myeloma. Blood 2021, 138, 2821. [Google Scholar] [CrossRef]
- Kumar, S.K.; Baz, R.C.; Orlowski, R.Z.; Anderson, L.D.; Ma, H.; Shrewsbury, A.; Croghan, K.A.; Bilgi, M.; Kansagra, A.; Kapoor, P.; et al. Results from Lummicar-2: A Phase 1b/2 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-Shelf” Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R.; Oluwole, O.O.; Karski, E.E.; Lovelace, W.; Zhou, X.; et al. Universal: An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Valton, J.; Galetto, R.; Bentley, T.; Sutton, J.; Ni, Y.; Leonard, M.; Van Blarcom, T.; Smith, J.; et al. ALLO-715, an Allogeneic BCMA CAR T Therapy Possessing an Off-Switch for the Treatment of Multiple Myeloma. Blood 2018, 132, 591. [Google Scholar] [CrossRef]
- Mailankody, S.; Liedtke, M.; Sidana, S.; Matous, J.V.; Chhabra, S.; Oluwole, O.O.; Malik, S.A.; Kumar, S.; Nath, R.; Anwer, F.; et al. Universal Updated Phase 1 Data Validates the Feasibility of Allogeneic Anti-BCMA ALLO-715 Therapy for Relapsed/Refractory Multiple Myeloma. Blood 2021, 138, 651. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Matthys, P.; Dillen, C.; Proost, P.; Heremans, H.; And, J.V.D.; Billiau, A. Modification of the Anti-CD3-Induced Cytokine Release Syndrome by Anti-Interferon-γ or Anti-Interleukin-6 Antibody Treatment: Protective Effects and Biphasic Changes in Blood Cytokine Levels. Eur. J. Immunol. 1993, 23, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Maus, M.V.; Alexander, S.; Bishop, M.R.; Brudno, J.N.; Callahan, C.; Davila, M.L.; Diamonte, C.; Dietrich, J.; Fitzgerald, J.C.; Frigault, M.J.; et al. Society for Immunotherapy of Cancer (SITC) Clinical Practice Guideline on Immune Effector Cell-Related Adverse Events. J. Immunother. Cancer 2020, 8, e001511. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Karschnia, P.; Jordan, J.T.; Forst, D.A.; Arrillaga-Romany, I.C.; Batchelor, T.T.; Baehring, J.M.; Clement, N.F.; Gonzalez Castro, L.N.; Herlopian, A.; Maus, M.V.; et al. Clinical Presentation, Management, and Biomarkers of Neurotoxicity after Adoptive Immunotherapy with CAR T Cells. Blood 2019, 133, 2212–2221. [Google Scholar] [CrossRef]
- Cohen, A.D.; Parekh, S.; Santomasso, B.D.; Pérez-Larraya, J.G.; van de Donk, N.W.C.J.; Arnulf, B.; Mateos, M.-V.; Lendvai, N.; Jackson, C.C.; De Braganca, K.C.; et al. Incidence and Management of CAR-T Neurotoxicity in Patients with Multiple Myeloma Treated with Ciltacabtagene Autoleucel in CARTITUDE Studies. Blood Cancer J. 2022, 12, 32. [Google Scholar] [CrossRef]
- Van Oekelen, O.; Aleman, A.; Upadhyaya, B.; Schnakenberg, S.; Madduri, D.; Gavane, S.; Teruya-Feldstein, J.; Crary, J.F.; Fowkes, M.E.; Stacy, C.B.; et al. Neurocognitive and Hypokinetic Movement Disorder with Features of Parkinsonism after BCMA-Targeting CAR-T Cell Therapy. Nat. Med. 2021, 27, 2099–2103. [Google Scholar] [CrossRef]
- Bu, D.-X.; Singh, R.; Choi, E.E.; Ruella, M.; Nunez-Cruz, S.; Mansfield, K.G.; Bennett, P.; Barton, N.; Wu, Q.; Zhang, J.; et al. Pre-Clinical Validation of B Cell Maturation Antigen (BCMA) as a Target for T Cell Immunotherapy of Multiple Myeloma. Oncotarget 2018, 9, 25764–25780. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and Management in CAR T-Cell Therapy. Mol. Ther.-Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.S.A.; Hsieh, C.-S. Development of T-Cell Tolerance Utilizes Both Cell-Autonomous and Cooperative Presentation of Self-Antigen. Immunol. Rev. 2016, 271, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive Immunotherapy for Cancer: Harnessing the T Cell Response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; de Witte, M.A.; Jorritsma, A.; Kaiser, A.D.M.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal Graft-versus-Host Disease in Mouse Models of T Cell Receptor Gene Therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef]
- Andrade, V.C.C.; Vettore, A.L.; Felix, R.S.; Almeida, M.S.S.; Carvalho, F.; Oliveira, J.S.R.; Chauffaille, M.L.L.F.; Andriolo, A.; Caballero, O.L.; Zago, M.A.; et al. Prognostic Impact of Cancer/Testis Antigen Expression in Advanced Stage Multiple Myeloma Patients. Cancer Immunol. 2008, 8, 2. [Google Scholar] [CrossRef]
- van Baren, N.; Brasseur, F.; Godelaine, D.; Hames, G.; Ferrant, A.; Lehmann, F.; André, M.; Ravoet, C.; Doyen, C.; Spagnoli, G.C.; et al. Genes Encoding Tumor-Specific Antigens Are Expressed in Human Myeloma Cells. Blood 1999, 94, 1156–1164. [Google Scholar]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–Specific TCR–Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, 6481. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Faitg, T.H.; Lowther, D.E.; Badros, A.Z.; Chagin, K.; Dengel, K.; Iyengar, M.; Melchiori, L.; Navenot, J.M.; Norry, E.; et al. Long-term Safety and Activity of NY-ESO-1 SPEAR T Cells after Autologous Stem Cell Transplant for Myeloma. Blood Adv. 2019, 3, 2022–2034. [Google Scholar] [CrossRef]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T Cell Specificity towards Leukemia by Zinc Finger Nucleases and Lentiviral Gene Transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Schober, K.; Müller, T.R.; Gökmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic Replacement of T-Cell Receptor α- and β-Chains with Preservation of near-Physiological T-Cell Function. Nat. Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Delrow, J.J.; Basom, R.S.; Blattman, J.N.; Greenberg, P.D. Rescued Tolerant CD8 T Cells Are Preprogrammed to Reestablish the Tolerant State. Science 2012, 335, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-Cell Receptor: Long-Term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, T.S.; Berent-Maoz, B.; Cheung-Lau, G.; Huang, R.R.; Wang, X.; Tsoi, J.; Kaplan-Lefko, P.; Cabrera, P.; Tran, J.; Pang, J.; et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-Reactive Adoptive Cellular Therapy with Dendritic Cell Vaccination with or without Ipilimumab. Clin. Cancer Res. 2019, 25, 2096–2108. [Google Scholar] [CrossRef]
- Mackall, C.; D’Angelo, S.P.; Grupp, S.A.; Odunsi, K.; Cristea, M.C.; Linette, G.P.; Kluger, H.M.; Kari, G.; Pandite, L.; Holdich, T.; et al. Autologous Genetically Engineered NY-ESO-1 C259 T in HLA-A*02:01, HLA*02:05 and HLA*02:06 Positive Patients with NY-ESO-1 Expressing Tumors. J. Clin. Oncol. 2016, 34, TPS3101. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T Cell Therapy Using Antigen-Specific CD8 + T Cell Clones for the Treatment of Patients with Metastatic Melanoma: In Vivo Persistence, Migration, and Antitumor Effect of Transferred T Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hont, A.B.; Cruz, C.R.; Ulrey, R.; O’Brien, B.; Stanojevic, M.; Datar, A.; Albihani, S.; Saunders, D.; Hanajiri, R.; Panchapakesan, K.; et al. Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J. Clin. Oncol. 2019, 37, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Lulla, P.D.; Tzannou, I.; Vasileiou, S.; Carrum, G.; Ramos, C.A.; Kamble, R.; Wang, T.; Wu, M.; Bilgi, M.; Gee, A.P.; et al. The Safety and Clinical Effects of Administering a Multiantigen-Targeted T Cell Therapy to Patients with Multiple Myeloma. Sci. Transl. Med. 2020, 12, 554. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.D.; Htut, M.; Varela, J.C.; Kin, A.; Edavana, V.; Lu, E.; Kim, S.; Suarez, L.; Oelke, M.; Bednarik, D. Phase 1/2 Study of Nexi-002 Autologous Multi-Antigen-Specific CD8+ T Cells for the Treatment of Relapsed or Refractory Multiple Myeloma. Blood 2021, 138, 2824. [Google Scholar] [CrossRef]
- Vasileiou, S.; Lulla, P.D.; Tzannou, I.; Watanabe, A.; Kuvalekar, M.; Callejas, W.L.; Bilgi, M.; Wang, T.; Wu, M.J.; Kamble, R.; et al. T-Cell Therapy for Lymphoma Using Nonengineered Multiantigen-Targeted T Cells Is Safe and Produces Durable Clinical Effects. J. Clin. Oncol. 2021, 39, 1415–1425. [Google Scholar] [CrossRef]
- Balermpas, P.; Rödel, F.; Rödel, C.; Krause, M.; Linge, A.; Lohaus, F.; Baumann, M.; Tinhofer, I.; Budach, V.; Gkika, E.; et al. CD8+ Tumour-Infiltrating Lymphocytes in Relation to HPV Status and Clinical Outcome in Patients with Head and Neck Cancer after Postoperative Chemoradiotherapy: A Multicentre Study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Int. J. Cancer 2016, 138, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Campbell, C.; Savas, P.; Nuciforo, P.; Aura, C.; De Azambuja, E.; Eidtmann, H.; Ellis, C.E.; Baselga, J.; et al. Tumor-Infiltrating Lymphocytes and Associations with Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated with Lapatinib and Trastuzumab: A Secondary Analysis of the NeoALTTO Trial. JAMA Oncol. 2015, 1, 448–455. [Google Scholar] [CrossRef]
- Kovacsovics-Bankowski, M.; Chisholm, L.; Vercellini, J.; Tucker, C.G.; Montler, R.; Haley, D.; Newell, P.; Ma, J.; Tseng, P.; Wolf, R.; et al. Detailed Characterization of Tumor Infiltrating Lymphocytes in Two Distinct Human Solid Malignancies Show Phenotypic Similarities. J. Immunother. Cancer 2014, 2, 38. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Li, J.; Chen, Q.Y.; He, J.; Li, Z.L.; Tang, X.F.; Chen, S.P.; Xie, C.M.; Li, Y.Q.; Huang, L.X.; Ye, S.B.; et al. Phase I Trial of Adoptively Transferred Tumor-Infiltrating Lymphocyte Immunotherapy Following Concurrent Chemoradiotherapy in Patients with Locoregionally Advanced Nasopharyngeal Carcinoma. Oncoimmunology 2015, 4, e976507. [Google Scholar] [CrossRef]
- Melioli, G.; Ratto, G.B.; Ponte, M.; Guastella, M.; Semino, C.; Fantino, G.; Tassara, E.; Pasquetti, W.; Mereu, C.; Merlo, F.; et al. Treatment of Stage IIIB Non-Small-Cell Lung Cancer with Surgery Followed by Infusion of Tumor Infiltrating Lymphocytes and Recombinant Interleukin-2: A Pilot Study. J. Immunother. 1996, 19, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive Transfer of Activated Marrow-Infiltrating Lymphocytes Induces Measurable Antitumor Immunity in the Bone Marrow in Multiple Myeloma. Sci. Transl. Med. 2015, 7, 288ra78. [Google Scholar] [CrossRef]
- Noonan, K.; Matsui, W.; Serafini, P.; Carbley, R.; Tan, G.; Khalili, J.; Bonyhadi, M.; Levitsky, H.; Whartenby, K.; Borrello, I. Activated Marrow-Infiltrating Lymphocytes Effectively Target Plasma Cells and Their Clonogenic Precursors. Cancer Res. 2005, 65, 2026–2034. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.H.; Yan, M.; Heuijerjans, J.; Carter, L.; Abolhassani, A.; Frosch, J.; Wallace, R.; Flutter, B.; Capsomidis, A.; Hubank, M.; et al. Neuroblastoma Killing Properties of Vδ2 and Vδ2-Negative ΓδT Cells Following Expansion by Artificial Antigen-Presenting Cells. Clin. Cancer Res. 2014, 20, 5720–5732. [Google Scholar] [CrossRef]
- Kabelitz, D.; Wesch, D.; Pitters, E.; Zöller, M. Potential of Human Γδ T Lymphocytes for Immunotherapy of Cancer. Int. J. Cancer 2004, 112, 727–732. [Google Scholar] [CrossRef]
- Hintz, M.; Reichenberg, A.; Altincicek, B.; Bahr, U.; Gschwind, R.M.; Kollas, A.-K.; Beck, E.; Wiesner, J.; Eberl, M.; Jomaa, H. Identification of ( E )-4-Hydroxy-3-Methyl-but-2-Enyl Pyrophosphate as a Major Activator for Human Γδ T Cells in Escherichia Coli. FEBS Lett. 2001, 509, 317–322. [Google Scholar] [CrossRef]
- Bürk, M.R.; Mori, L.; de Libero, G. Human Vγ9-Vδ2 Cells Are Stimulated in a Crossreactive Fashion by a Variety of Phosphorylated Metabolites. Eur. J. Immunol. 1995, 25, 2052–2058. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Weissinger, F.; Tony, H.P.; Wilhelm, M. Stimulation of Γδ T Cells by Aminobisphosphonates and Induction of Antiplasma Cell Activity in Multiple Myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Presti, E.L.; Pizzolato, G.; Gulotta, E.; Cocorullo, G.; Gulotta, G.; Dieli, F.; Meraviglia, S. Current Advances in Γδ T Cell-Based Tumor Immunotherapy. Front. Immunol. 2017, 8, 1401. [Google Scholar] [CrossRef]
- Gertner-Dardenne, J.; Bonnafous, C.; Bezombes, C.; Capietto, A.-H.; Scaglione, V.; Ingoure, S.; Cendron, D.; Gross, E.; Lepage, J.-F.; Quillet-Mary, A.; et al. Bromohydrin Pyrophosphate Enhances Antibody-Dependent Cell-Mediated Cytotoxicity Induced by Therapeutic Antibodies. Blood 2009, 113, 4875–4884. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional Antigen-Presentation Function by Human Γδ T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, S.; Lo Presti, E.; Tosolini, M.; La Mendola, C.; Orlando, V.; Todaro, M.; Catalano, V.; Stassi, G.; Cicero, G.; Vieni, S.; et al. Distinctive Features of Tumor-Infiltrating Γδ T Lymphocytes in Human Colorectal Cancer. Oncoimmunology 2017, 6, e1347742. [Google Scholar] [CrossRef] [PubMed]
- Burjanadzé, M.; Condomines, M.; Reme, T.; Quittet, P.; Latry, P.; Lugagne, C.; Romagne, F.; Morel, Y.; Rossi, J.F.; Klein, B.; et al. In Vitro Expansion of Gamma Delta T Cells with Anti-Myeloma Cell Activity by Phosphostim and IL-2 in Patients with Multiple Myeloma. Br. J. Haematol. 2007, 139, 206–216. [Google Scholar] [CrossRef]
- Castella, B.; Vitale, C.; Coscia, M.; Massaia, M. Vγ9Vδ2 T Cell-Based Immunotherapy in Hematological Malignancies: From Bench to Bedside. Cell. Mol. Life Sci. 2011, 68, 2419–2432. [Google Scholar] [CrossRef]
- Castella, B.; Foglietta, M.; Sciancalepore, P.; Rigoni, M.; Coscia, M.; Griggio, V.; Vitale, C.; Ferracini, R.; Saraci, E.; Omedé, P.; et al. Anergic Bone Marrow Vγ9Vδ2 T Cells as Early and Long-Lasting Markers of PD-1-Targetable Microenvironment-Induced Immune Suppression in Human Myeloma. Oncoimmunology 2015, 4, e1047580. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Castella, B.; Foglietta, M.; Riganti, C.; Massaia, M. Vγ9Vδ2 T Cells in the Bone Marrow of Myeloma Patients: A Paradigm of Microenvironment-Induced Immune Suppression. Front. Immunol. 2018, 9, 1492. [Google Scholar] [CrossRef]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and Immunological Evaluation of Zoledronate-Activated Vγ9γδ T-Cell-Based Immunotherapy for Patients with Multiple Myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Product | Phase | Specificity | scFv Binding Protein | Median Lines of Therapy | Dose | Response Rates (ORR/CR) | m-PFS | CRS (all/Gr ≥ 3) | ICANS (all/Gr ≥ 3) |
|---|---|---|---|---|---|---|---|---|---|
| Ide-cel [14] (n = 128) | II | Autologous | Murine | 6 | 150–450 mil | 73%/33% | 8.8 months | 84%/5% | 18/3% |
| Cilta-cel [15,17] (n = 97) | Ib/II | Autologous | Camelid | 6 | 0.75 mil/Kg | 97.9%/82.5% | NR | 95%/5% | 17%/2% |
| bb21217 [32] (n = 72) | I | Autologous | Murine | 6 | 150–450 mil | 69%/28% | Not reported | 75%/4% | 15%/4% |
| Orva-cel [42] (n = 51) | I/II | Autologous | Human | 6 | 300–600 mil | 91%/39% | Not reported | 2% (Gr ≥ 3) | 4% (Gr ≥ 3) |
| Zevor-cel [47] (n = 20) | I | Autologous | Human | 6 | 150–300 mil | 94%/28% | Not reported | 86%/0% | 7%/0% |
| ALLO-715 (n = 42) | I | Allogeneic | Human | 5 | 40–480 mil | 61.5%/38.5%* | Not reported | 52.4%/2% | 2%/0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simmons, G.L.; Castaneda Puglianini, O. T-Cell-Based Cellular Immunotherapy of Multiple Myeloma: Current Developments. Cancers 2022, 14, 4249. https://doi.org/10.3390/cancers14174249
Simmons GL, Castaneda Puglianini O. T-Cell-Based Cellular Immunotherapy of Multiple Myeloma: Current Developments. Cancers. 2022; 14(17):4249. https://doi.org/10.3390/cancers14174249
Chicago/Turabian StyleSimmons, Gary L., and Omar Castaneda Puglianini. 2022. "T-Cell-Based Cellular Immunotherapy of Multiple Myeloma: Current Developments" Cancers 14, no. 17: 4249. https://doi.org/10.3390/cancers14174249