
Whole-Exome Sequencing Reveals the Genomic Features of the Micropapillary Component in Ground-Glass Opacities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Microdissection
2.2. WES
2.3. Oncogenic Signaling Pathway
2.4. Analysis of the Copy Number Variation
2.5. Calculation of the dN/dS Value
2.6. Statistical Analysis
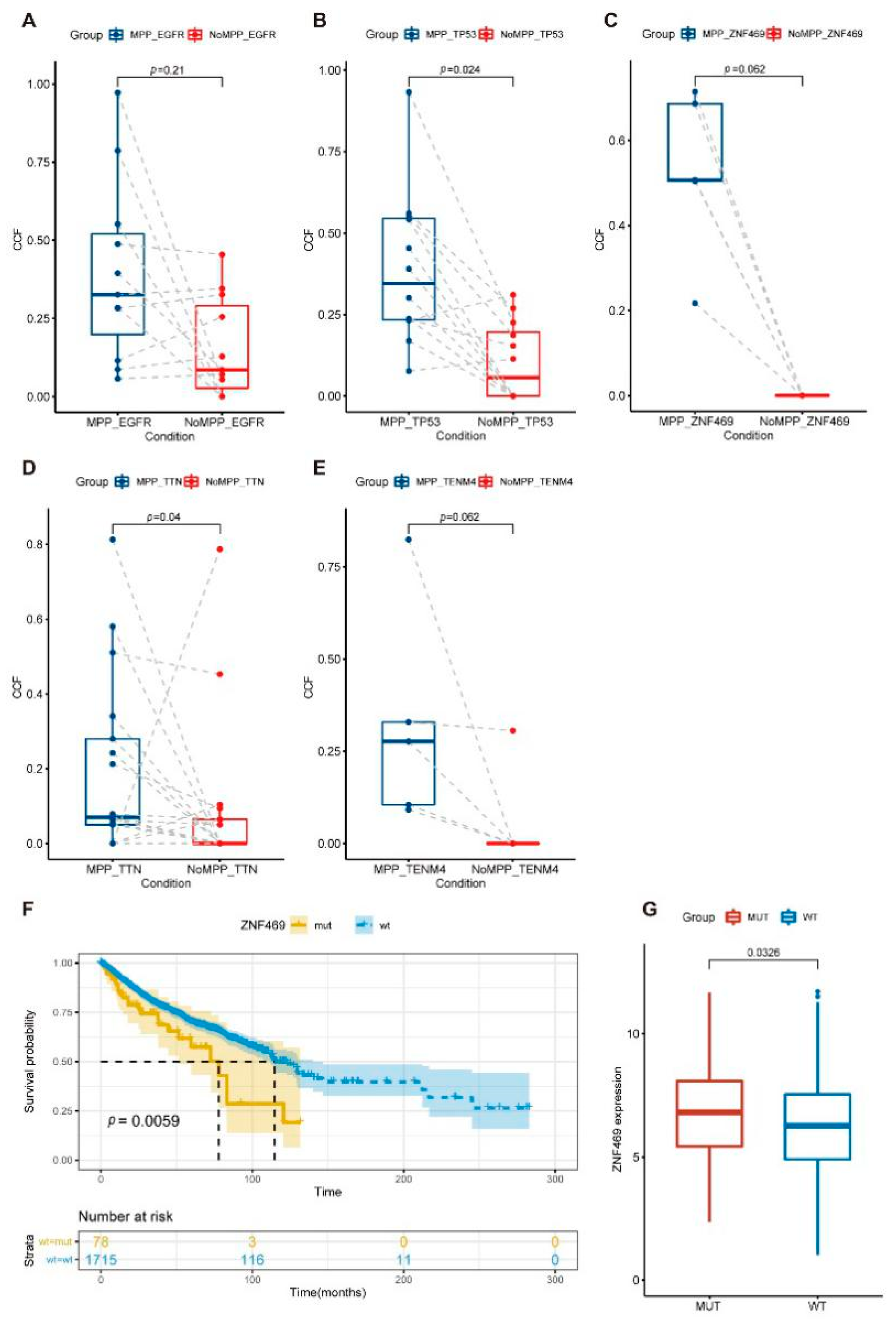
3. Results
3.1. Study Design
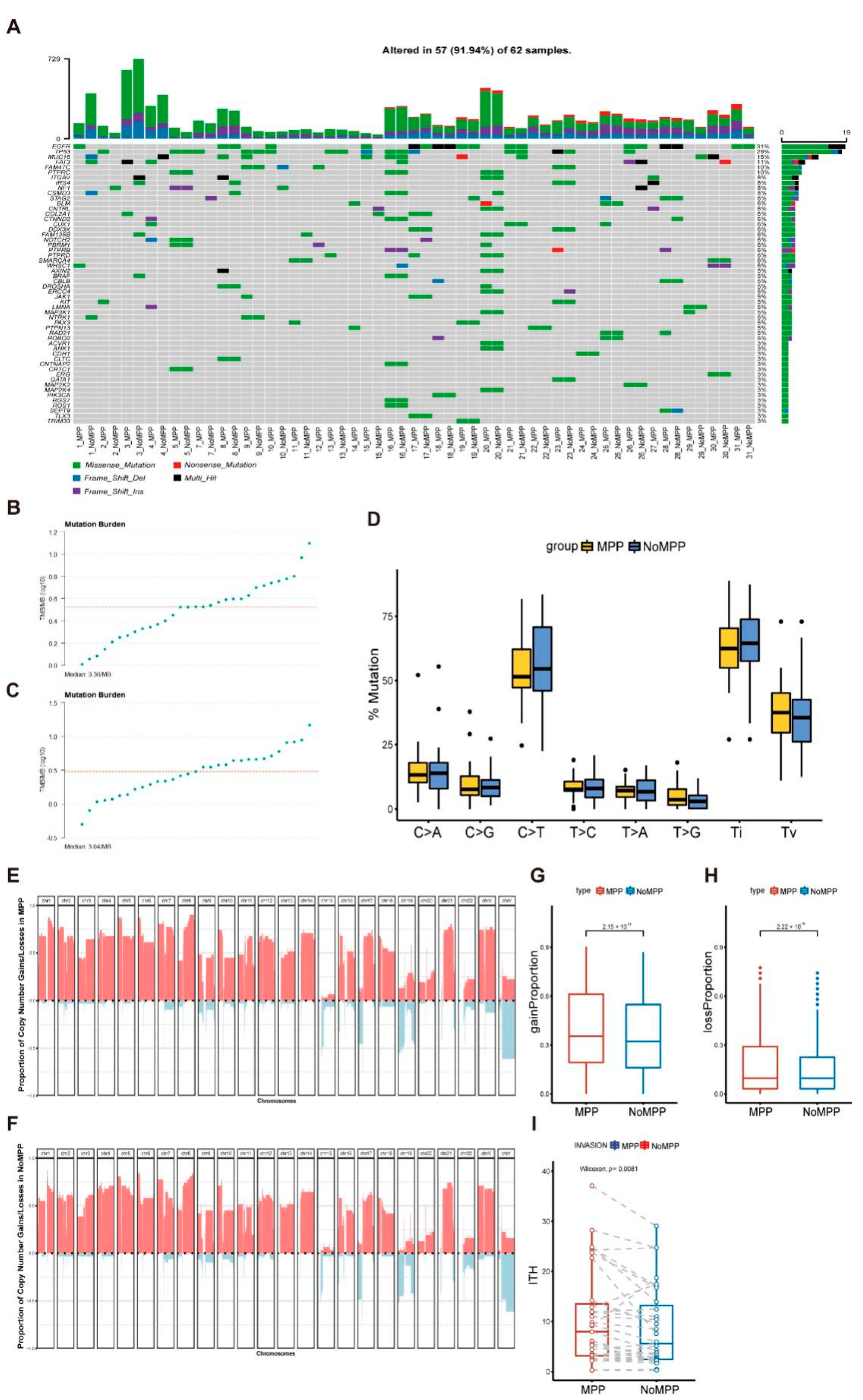
3.2. Mutational Landscape in the MPP Component of GGOs
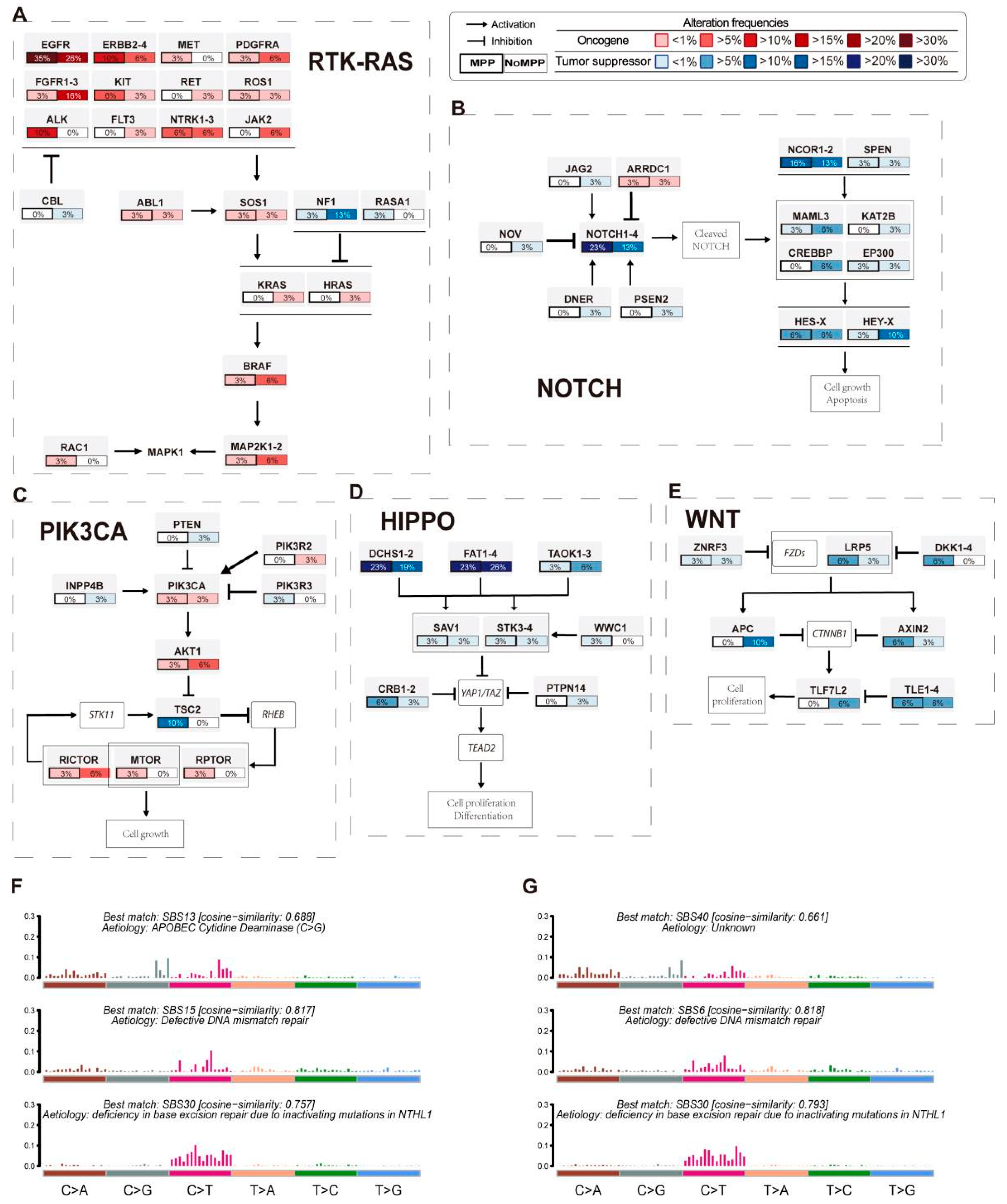
3.3. Analysis of Mutation Signatures and Oncogenic Pathways in the MPP Component of GGOs
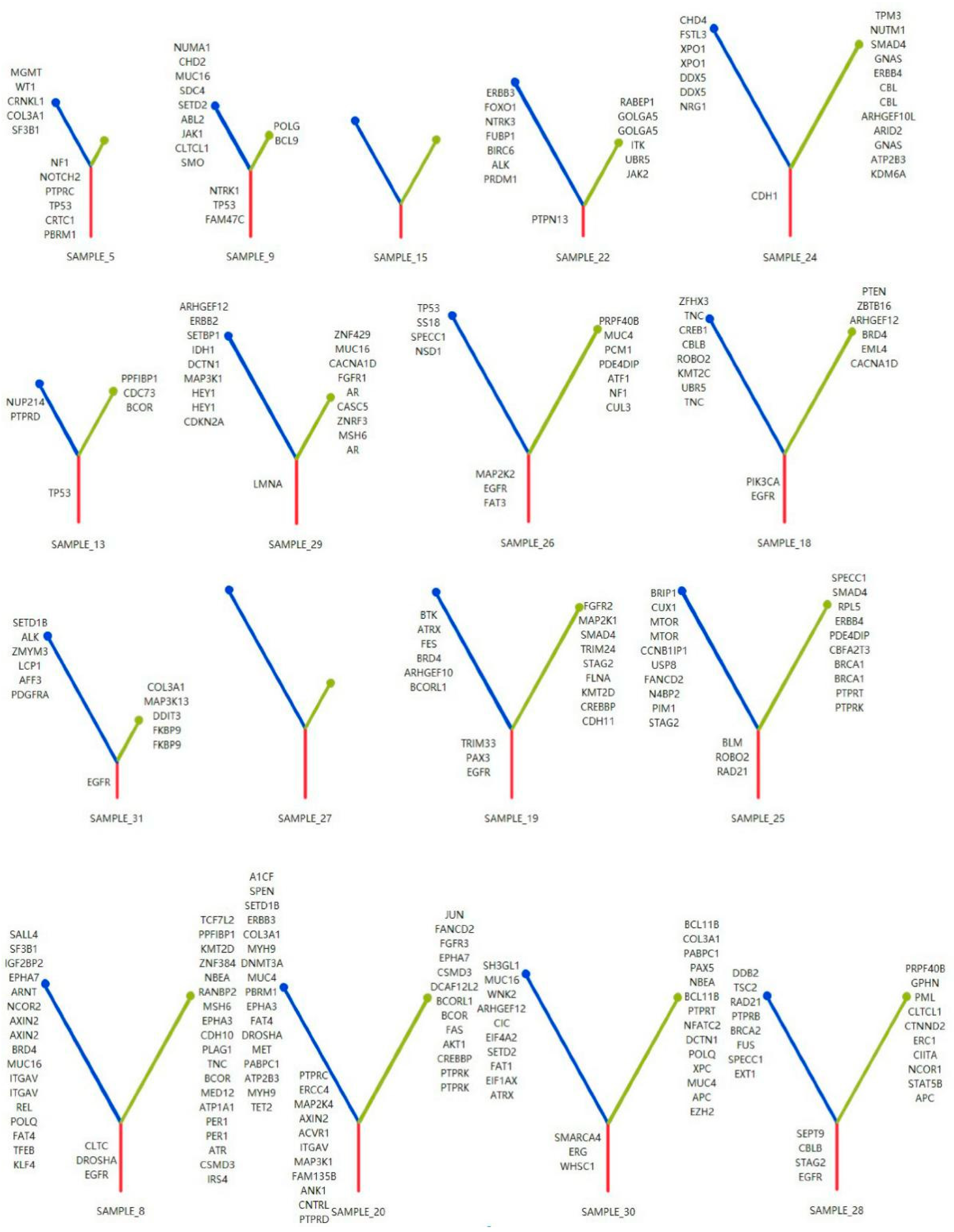
3.4. Phylogenetic Analysis of GGOs Associated with Mutations in the MPP Component
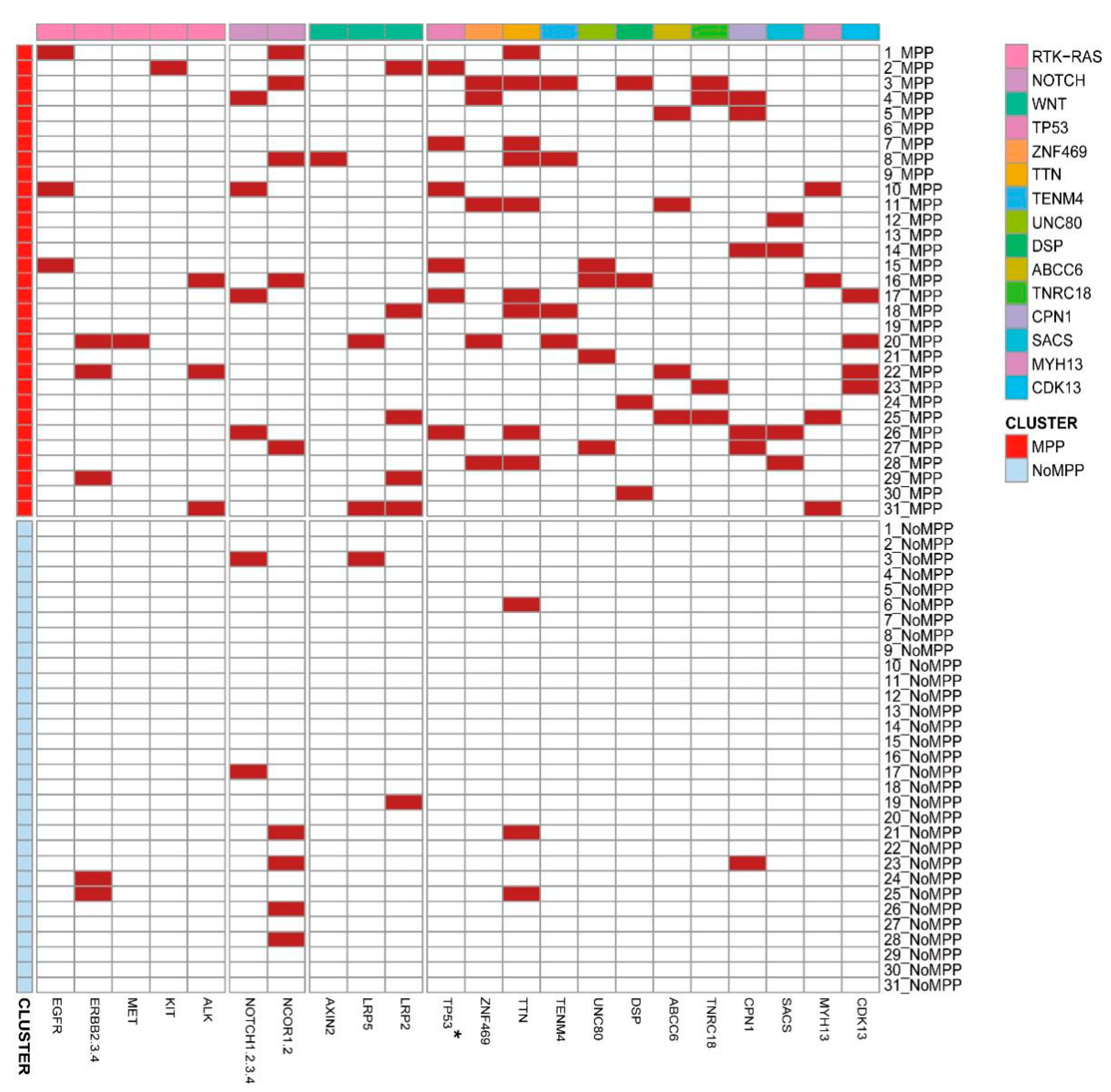
3.5. Distribution of Driver Mutations and Selection Pressure during the Evolution of MPP Components in GGOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Jones, G.D.; Brandt, W.S.; Shen, R.; Sanchez-Vega, F.; Tan, K.S.; Martin, A.; Jones, D.R. A Genomic-Pathologic Annotated Risk Model to Predict Recurrence in Early-Stage Lung Adenocarcinoma. JAMA Surg. 2021, 156, e205601. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Wistuba, I. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fan, Y.; Lang, R.G.; Guo, X.J.; Sun, Y.L.; Cui, L.F.; Fu, L. Breast carcinoma with micropapillary features: Clinicopathologic study and long-term follow-up of 100 cases. Int. J. Surg. Pathol. 2008, 16, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Han, Y.; Wang, C.; Guo, X.; Shen, B.; Liu, F.; Fu, L. Precise pathologic diagnosis and individualized treatment improve the outcomes of invasive micropapillary carcinoma of the breast: A 12-year prospective clinical study. Mod Pathol. 2018, 31, 956–964. [Google Scholar] [CrossRef]
- Luna-Moré, S.; Gonzalez, B.; Acedo, C.; Rodrigo, I.; Luna, C. Invasive micropapillary carcinoma of the breast. A new special type of invasive mammary carcinoma. Pathol. Res. Pract. 1994, 190, 668–674. [Google Scholar] [CrossRef]
- Guo, X.; Chen, L.; Lang, R.; Fan, Y.; Zhang, X.; Fu, L. Invasive micropapillary carcinoma of the breast: Association of pathologic features with lymph node metastasis. Am. J. Clin. Pathol. 2006, 126, 740–746. [Google Scholar] [CrossRef]
- Nassar, H. Carcinomas with micropapillary morphology: Clinical significance and current concepts. Adv. Anat. Pathol. 2004, 11, 297–303. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R.; Cai, D.; Li, Y.; Pan, Y.; Hu, H.; Chen, H. A comprehensive investigation of molecular features and prognosis of lung adenocarcinoma with micropapillary component. J. Thorac. Oncol. 2014, 9, 1772–1778. [Google Scholar] [CrossRef]
- Warth, A.; Penzel, R.; Lindenmaier, H.; Brandt, R.; Stenzinger, A.; Herpel, E.; Weichert, W. EGFR, KRAS, BRAF and ALK gene alterations in lung adenocarcinomas: Patient outcome, interplay with morphology and immunophenotype. Eur. Respir. J. 2014, 43, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Caso, R.; Sanchez-Vega, F.; Tan, K.S.; Mastrogiacomo, B.; Zhou, J.; Jones, G.D.; Jones, D.R. The Underlying Tumor Genomics of Predominant Histologic Subtypes in Lung Adenocarcinoma. J. Thorac. Oncol. 2020, 15, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Mroz, E.A.; Rocco, J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral. Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Shi, Q.; Shao, K.; Jia, H.; Cao, B.; Li, W.; Dong, S.; Fu, L. Genomic alterations and evolution of cell clusters in metastatic invasive micropapillary carcinoma of the breast. Nat. Commun. 2022, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Shi, Q.; Han, Y.; Li, W.; Liu, H.; Zhang, J.; Fu, L. Spatial transcriptomics reveals gene expression characteristics in invasive micropapillary carcinoma of the breast. Cell Death Dis. 2021, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Zhai, W. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Stratton, M.R. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Stratton, M.R. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Futreal, P.A. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Y.; Zhao, P.; Bao, H.; Wang, X.; Liu, R.; Ma, S. Integrated Analysis of Genomic and Immunological Features in Lung Adenocarcinoma with Micropapillary Component. Front. Oncol. 2021, 11, 652193. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, L.; Xu, T.; Xue, R.; Yu, L.; Zhu, Y.; Zhang, H. Mapping the spreading routes of lymphatic metastases in human colorectal cancer. Nat. Commun. 2020, 11, 1993. [Google Scholar] [CrossRef]
- Wolfe, K.; Ó’hUigín, C. Significance of positive selection and gene duplication in adaptive evolution: In memory of Austin L. Hughes. Immunogenetics 2016, 68, 749–753. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e1021. [Google Scholar] [CrossRef]
- Strauss, G.M.; Kwiatkowski, D.J.; Harpole, D.H.; Lynch, T.J.; Skarin, A.T.; Sugarbaker, D.J. Molecular and pathologic markers in stage I non-small-cell carcinoma of the lung. J. Clin. Oncol. 1995, 13, 1265–1279. [Google Scholar] [CrossRef] [PubMed]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Beer, D.G. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Mountain, C.F. Staging classification of lung cancer. A critical evaluation. Clin. Chest Med. 2002, 23, 103–121. [Google Scholar] [CrossRef]
- Fidler, I.J. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer 1973, 9, 223–227. [Google Scholar] [CrossRef]
- Liotta, L.A.; Saidel, M.G.; Kleinerman, J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976, 36, 889–894. [Google Scholar]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Kishi, N.; Ito, M.; Miyata, Y.; Kanai, A.; Handa, Y.; Tsutani, Y.; Okada, M. Intense Expression of EGFR L858R Characterizes the Micropapillary Component and L858R Is Associated with the Risk of Recurrence in pN0M0 Lung Adenocarcinoma with the Micropapillary Component. Ann. Surg. Oncol. 2020, 27, 945–955. [Google Scholar] [CrossRef]
- Chahoud, J.; Gleber-Netto, F.O.; McCormick, B.Z.; Rao, P.; Lu, X.; Guo, M.; Pettaway, C.A. Whole-exome Sequencing in Penile Squamous Cell Carcinoma Uncovers Novel Prognostic Categorization and Drug Targets Similar to Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 2560–2570. [Google Scholar] [CrossRef]
- Takemoto, A.; Tanimoto, K.; Mori, S.; Inoue, J.; Fujiwara, N.; Noda, T.; Inazawa, J. Integrative genome-wide analyses reveal the transcriptional aberrations in Japanese esophageal squamous cell carcinoma. Cancer Sci. 2021, 112, 4377–4392. [Google Scholar] [CrossRef]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Mimori, K. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagné, A.; Enlow, W.; Pigeon, M.A.; Orain, M.; Turcotte, S.; Bossé, Y.; Joubert, P. Comprehensive Assessment of PD-L1 Staining Heterogeneity in Pulmonary Adenocarcinomas Using Tissue Microarrays: Impact of the Architecture Pattern and the Number of Cores. Am. J. Surg. Pathol. 2018, 42, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Lv, J.W.; Mao, Y.P.; Li, X.M.; Li, J.Y.; Wang, Y.Q.; Ma, J. Unraveling tumour microenvironment heterogeneity in nasopharyngeal carcinoma identifies biologically distinct immune subtypes predicting prognosis and immunotherapy responses. Mol. Cancer 2021, 20, 14. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, F.; Zhang, Y.; Wang, S.; Liu, T.; Sun, M.; Zhu, H.; Dong, G.; Xia, Z.; You, J.; Kong, X.; et al. Whole-Exome Sequencing Reveals the Genomic Features of the Micropapillary Component in Ground-Glass Opacities. Cancers 2022, 14, 4165. https://doi.org/10.3390/cancers14174165
Meng F, Zhang Y, Wang S, Liu T, Sun M, Zhu H, Dong G, Xia Z, You J, Kong X, et al. Whole-Exome Sequencing Reveals the Genomic Features of the Micropapillary Component in Ground-Glass Opacities. Cancers. 2022; 14(17):4165. https://doi.org/10.3390/cancers14174165
Chicago/Turabian StyleMeng, Fanchen, Yi Zhang, Siwei Wang, Tongyan Liu, Mengting Sun, Hongyu Zhu, Guozhang Dong, Zhijun Xia, Jing You, Xiangru Kong, and et al. 2022. "Whole-Exome Sequencing Reveals the Genomic Features of the Micropapillary Component in Ground-Glass Opacities" Cancers 14, no. 17: 4165. https://doi.org/10.3390/cancers14174165