
Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
2.1. Machine Learning Algorithms
2.2. Single-Cell Analysis
2.3. Bulk Sample Meta-Analysis
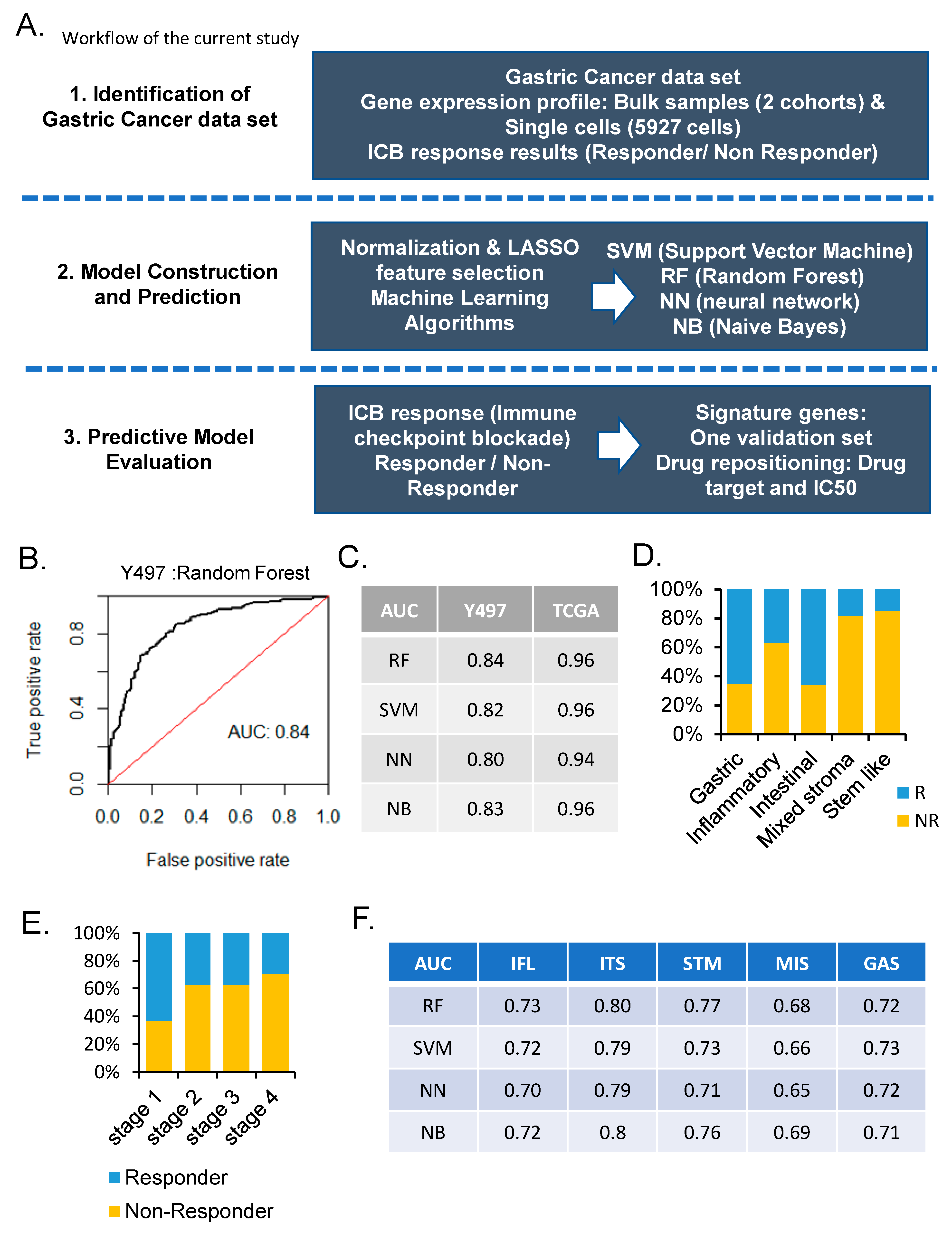
3. Results
3.1. Prediction of ICB Response of Machine Learning Algorithms
3.2. Prediction of ICB Response According to Single-Cell Types
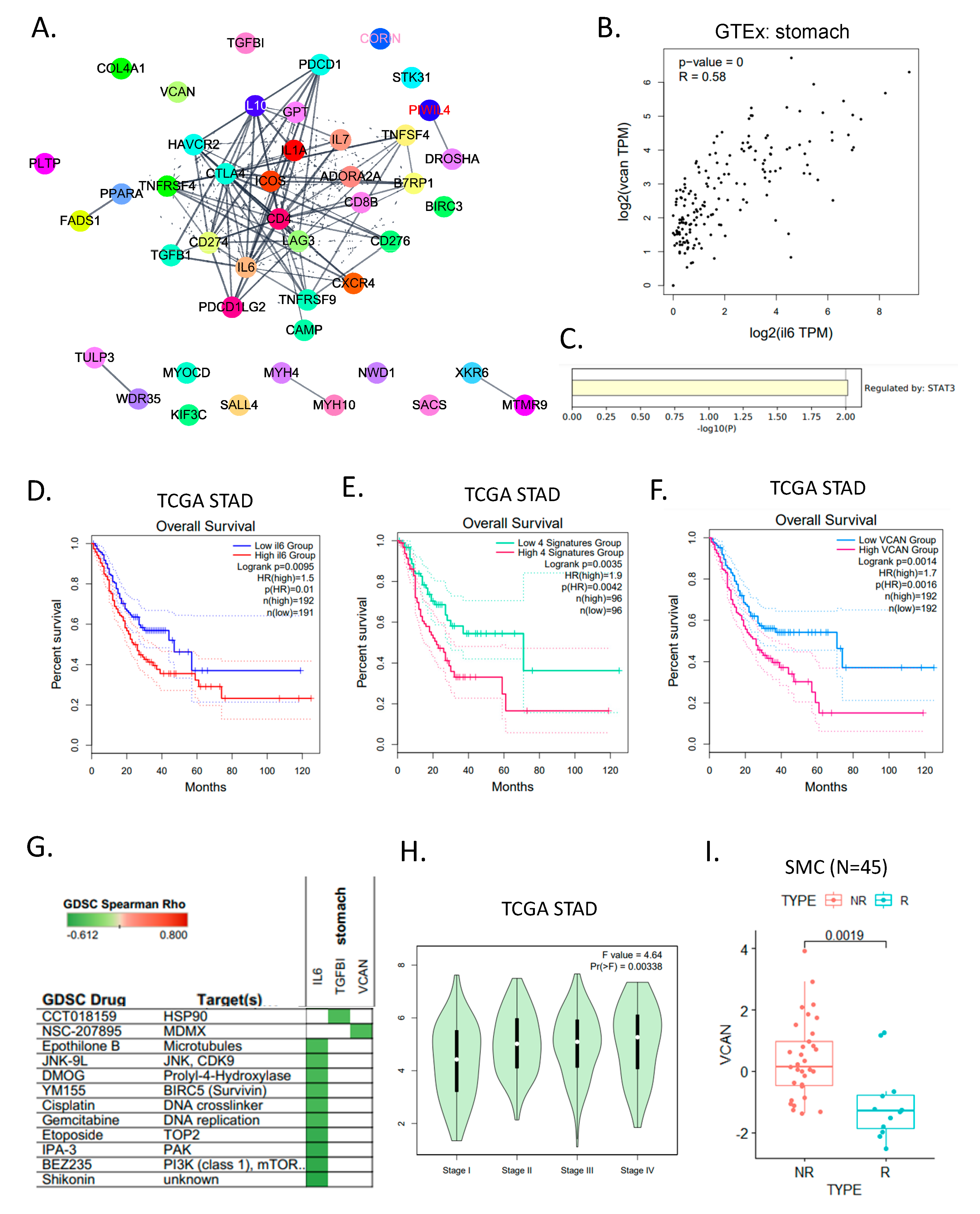
3.3. Molecular Characteristics of ICB Response According to Subtypes of Gastric Cancer
3.4. Evaluation and Validation of ICB Response Signature Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ICB | immune checkpoint blockade |
| STAD | stomach adenocarcinoma |
| PPI | protein–protein interaction |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| TCGA | The Cancer Genome Atlas |
| IGF | insulin-like growth factor |
| IL | interleukin |
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.-Y.; Cheong, J.-H. New Immunometabolic Strategy Based on Cell Type-Specific Metabolic Reprogramming in the Tumor Immune Microenvironment. Cells 2022, 11, 768. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Rihawi, K.; Ricci, A.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.; Llimpe, F.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Rizzo, A.; Llimpe, F.R.; Di Fabio, F.; De Biase, D.; Rihawi, K. Novel HER2-Directed Treatments in Advanced Gastric Carcinoma: AnotHER Paradigm Shift? Cancers 2021, 13, 1664. [Google Scholar] [CrossRef]
- Sammut, S.-J.; Crispin-Ortuzar, M.; Chin, S.-F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2021, 601, 623–629. [Google Scholar] [CrossRef]
- Cheong, J.-H.; Wang, S.C.; Park, S.; Porembka, M.R.; Christie, A.L.; Kim, H.; Kim, H.S.; Zhu, H.; Hyung, W.J.; Noh, S.H.; et al. Development and validation of a prognostic and predictive 32-gene signature for gastric cancer. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef]
- Sung, J.-Y.; Shin, H.-T.; Sohn, K.-A.; Shin, S.-Y.; Park, W.-Y.; Joung, J.-G. Assessment of intratumoral heterogeneity with mutations and gene expression profiles. PLoS ONE 2019, 14, e0219682. [Google Scholar] [CrossRef] [Green Version]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Kang, M.; Ko, E.; Mersha, T.B. A roadmap for multi-omics data integration using deep learning. Brief. Bioinform. 2021, 23, bbab454. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingos, P.; Pazzani, M. On the Optimality of the Simple Bayesian Classifier under Zero-One Loss. Mach. Learn. 1997, 29, 103–130. [Google Scholar] [CrossRef]
- Scholkopf, B.; Sung, K.-K.; Burges, C.; Girosi, F.; Niyogi, P.; Poggio, T.; Vapnik, V. Comparing support vector machines with Gaussian kernels to radial basis function classifiers. IEEE Trans. Signal. Process. 1997, 45, 2758–2765. [Google Scholar] [CrossRef] [Green Version]
- Hopfield, J.J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Kim, J.; Park, C.; Kim, K.H.; Kim, E.H.; Kim, H.; Woo, J.K.; Seong, J.K.; Nam, K.T.; Lee, Y.C.; Cho, S.Y. Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis. Oncol. 2022, 6, 1–11. [Google Scholar] [CrossRef]
- Van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729.e27. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Conley, A.P.; Grimm, E.A.; Roszik, J. A tool for discovering drug sensitivity and gene expression associations in cancer cells. PLoS ONE 2017, 12, e0176763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Cheong, J.-H.; Yang, H.-K.; Kim, H.; Kim, W.H.; Kim, Y.-W.; Kook, M.-C.; Park, Y.-K.; Kim, H.-H.; Lee, H.S.; Lee, K.H.; et al. Predictive test for chemotherapy response in resectable gastric cancer: A multi-cohort, retrospective analysis. Lancet Oncol. 2018, 19, 629–638. [Google Scholar] [CrossRef]
- Sung, J.-Y.; Cheong, J.-H. The Matrisome Is Associated with Metabolic Reprograming in Stem-like Phenotypes of Gastric Cancer. Cancers 2022, 14, 1438. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Yee, A.J. Roles of versican in cancer biology-tumorigenesis, progression and metastasis. Histol. Histopathol. 2013, 28, 701–713. [Google Scholar]
- Gao, R.; Cao, C.; Zhang, M.; Lopez, M.-C.; Yan, Y.; Chen, Z.; Mitani, Y.; Zhang, L.; Zajac-Kaye, M.; Liu, B.; et al. A unifying gene signature for adenoid cystic cancer identifies parallel MYB-dependent and MYB-independent therapeutic targets. Oncotarget 2014, 5, 12528–12542. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yee, A.J. Versican V2 isoform enhances angiogenesis by regulating endothelial cell activities and fibronectin expression. FEBS Lett. 2012, 587, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, K.; Karpova, M.; Asagoe, K.; Georgiev, O.; Dummer, R.; Urosevic-Maiwald, M. Versican upregulation in Sezary cells alters growth, motility and resistance to chemotherapy. Leukemia 2015, 29, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Cheong, J.H. Prognosis-related gene signature is enriched in cancer-associated fibroblasts in the stem-like subtype of gastric cancer. Clin. Transl. Med. 2022, 12, e930. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, J.-Y.; Cheong, J.-H. Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer. Cancers 2022, 14, 3191. https://doi.org/10.3390/cancers14133191
Sung J-Y, Cheong J-H. Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer. Cancers. 2022; 14(13):3191. https://doi.org/10.3390/cancers14133191
Chicago/Turabian StyleSung, Ji-Yong, and Jae-Ho Cheong. 2022. "Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer" Cancers 14, no. 13: 3191. https://doi.org/10.3390/cancers14133191