
Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer?


Abstract
:Simple Summary
Abstract
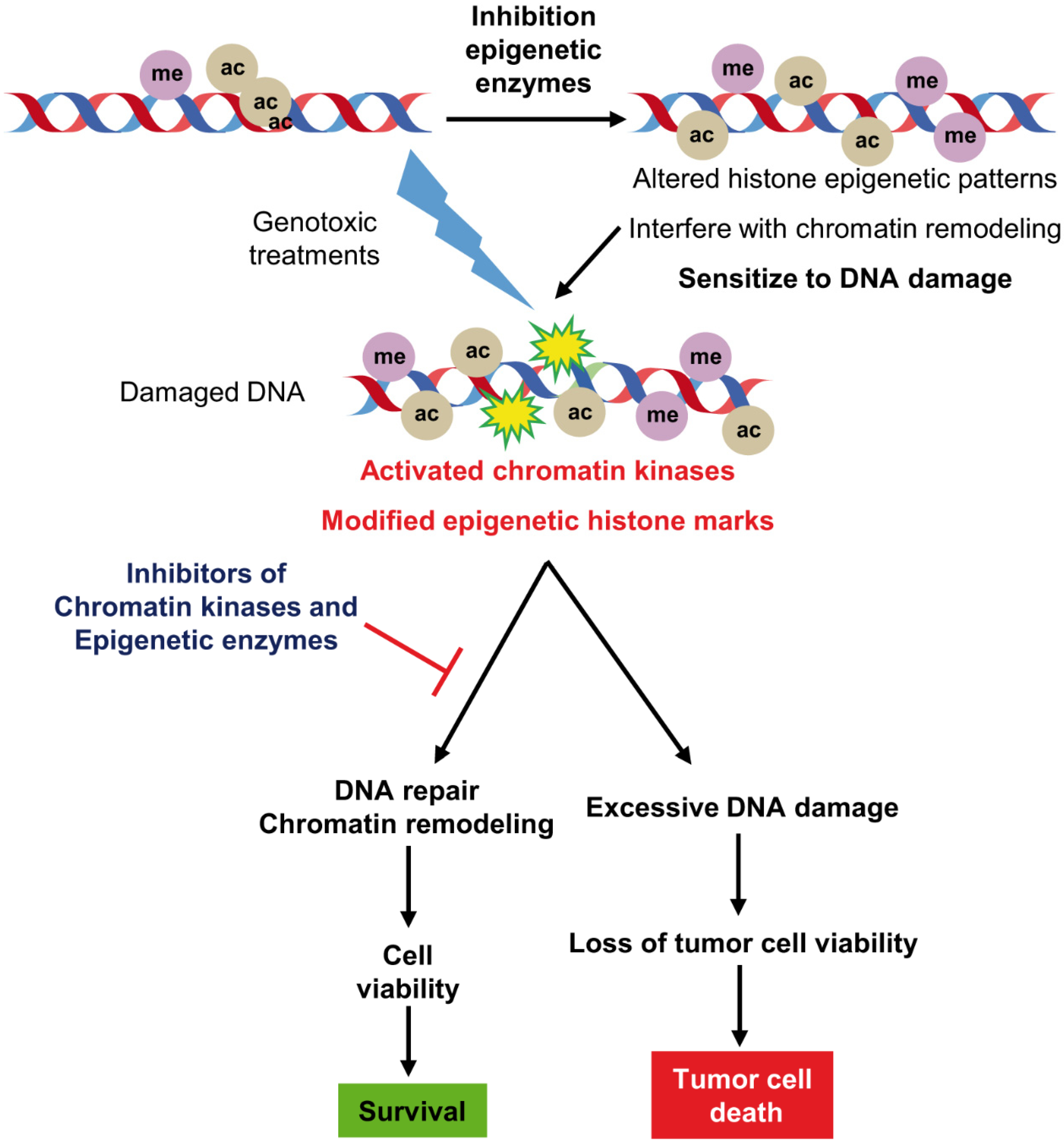
1. Introduction
2. Synthetic Lethality and Cancer
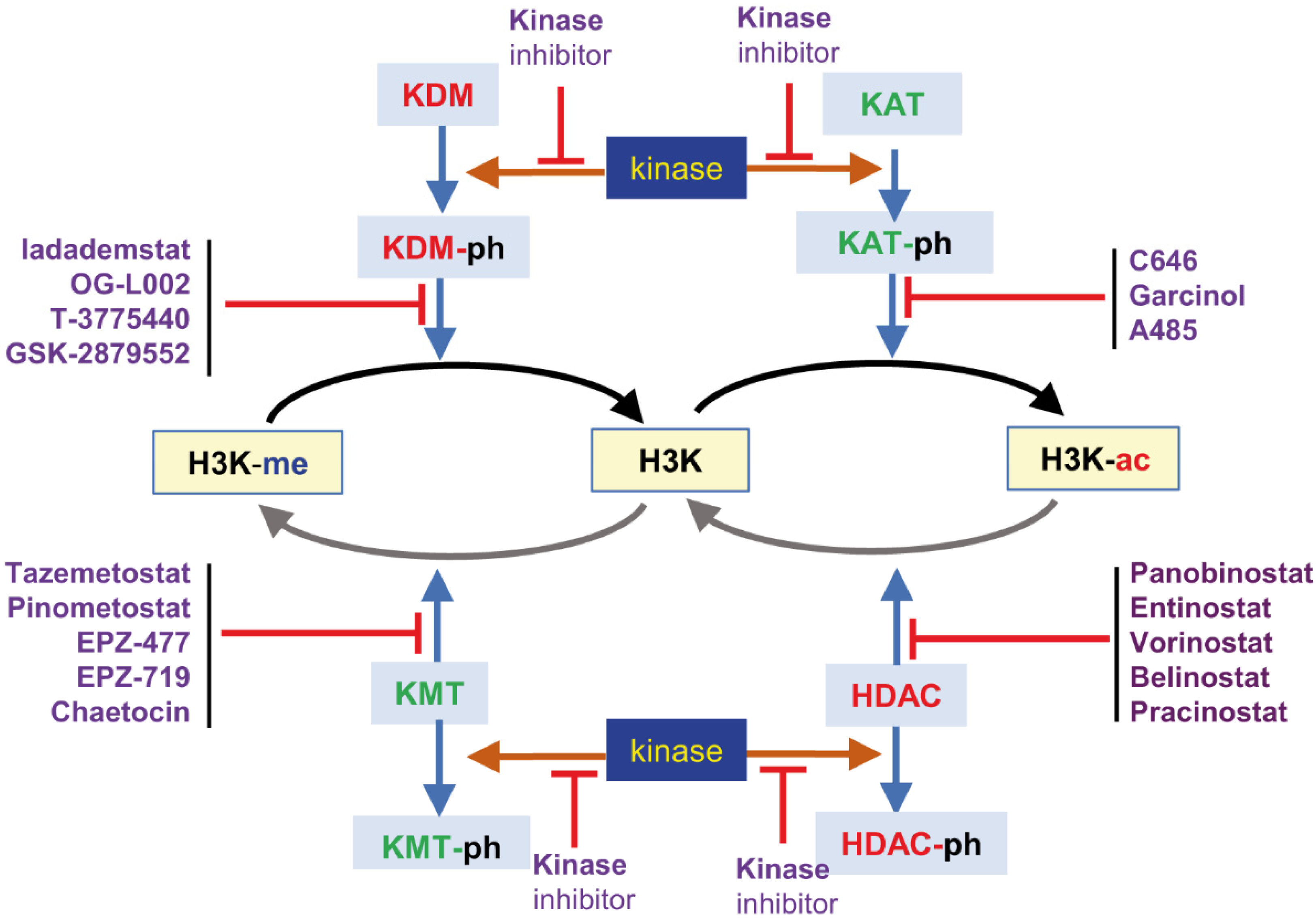
3. Epigenetic Chromatin Remodeling as Pharmacological Target
4. Histone Methylation
4.1. Targeting Lysine Methyl Transferases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Combinations | Inhibitor Combinations | Tumor | Ref. | |
|---|---|---|---|---|
| 1 | NSD2 (KMT3G) | SRC-3 | Multiple myeloma | [46] |
| Proteasome | bortezomid | |||
| 2 | NSD2 (KMT3G) | INCB054329 | Multiple myeloma | [62] |
| JAK1 | itacitinib | |||
| 3 | NSD2 (KMT3G) | Tazemetostat | Osteosarcoma, leiomyosarcoma cell lines | [33] |
| DNA intercalation/Topoisomerase I inhibition | Doxorubicin | |||
| 4 | EZH2 (KMT6A) | Tazemetostat | Osteosarcoma, leiomyosarcoma cell lines | [33] |
| DNMT | Azacitidine/Decitabine | |||
| 5 | DOT1L (KMT4) | Pinometostat (EPZ-5676) | MLL rearrangements | [29] |
| Topoisomerase I inhibitors, DNA methylation inhibitor | daunorubicin Azacytidine | [30] | ||
| 6 | DOT1L (KMT4) | EPZ-4777 | MLL rearrangements | [31] |
| Sirt1 | SRT1720 | |||
| 7 | SETD2 (KMT3A) | EPZ-719 | Preclinical | [53] |
| 8 | SETDB1 (KMT1E) | mithramycin | Preclinical | [53,57] |
| DNA methylation | Azacytidine | [54] | ||
| 9 | G9A (KMT1C) | HKMTI-1-005 | Melanoma, ovarian carcinoma | [59,60] |
| EZH2 | ||||
| 10 | KMT | chaetocin | NSCLC cell lines | [63] |
| DNA damage | IR, doxorubicin | |||
| 11 | KMT | chaetocin | Hepatoma and sarcoma cell lines | [33,64] |
| Autophagy/Atg5 | Bafilomycin A1 | |||
| 12 | KMT | Tazemetostat, chaetocin | Osteosarcoma, leiomyosarcoma lines | [33] |
| DNA damage | doxorubicin | |||
| 13 | LSD1 (KDM1A) | ORY-1001 (iadademstat) | AML | [65] |
| 14 | LSD1 (KDM1A) | ORY-1001 (iadademstat) | Luminal B and HER2 amplified breast cancer | [66] |
| 15 | LSD1 (KDM1A) | T-3775440 | Small cell lung carcinoma (SCLC) | [67] |
| 16 | LSD1 (KDM1A) | GSK2879552 | SCLC cell lines | [68,69] |
| 17 | LSD1 (KDM1A) | tranylcypromine GSK2879552 | Acute myeloid leukemia (AML) | [70,71] |
| MEK1 | trametinib | |||
| 18 | LSD1 (KDM6) | Corin dual inhibitor | diffuse intrinsic pontine glioma (DIPGs) | [72] |
| HDAC1A | ||||
4.2. Targeting Lysine Demethylases (KDM)
5. Targeting Histone Acetylases
5.1. Targeting Lysine Deacetylases
5.2. Targeting Lysine Acetyl Transferases (KAT)
6. Targeting Nuclear Kinases Associated with Chromatin Remodeling and Damage Response Pathways
6.1. Haspin and Aurora Kinases
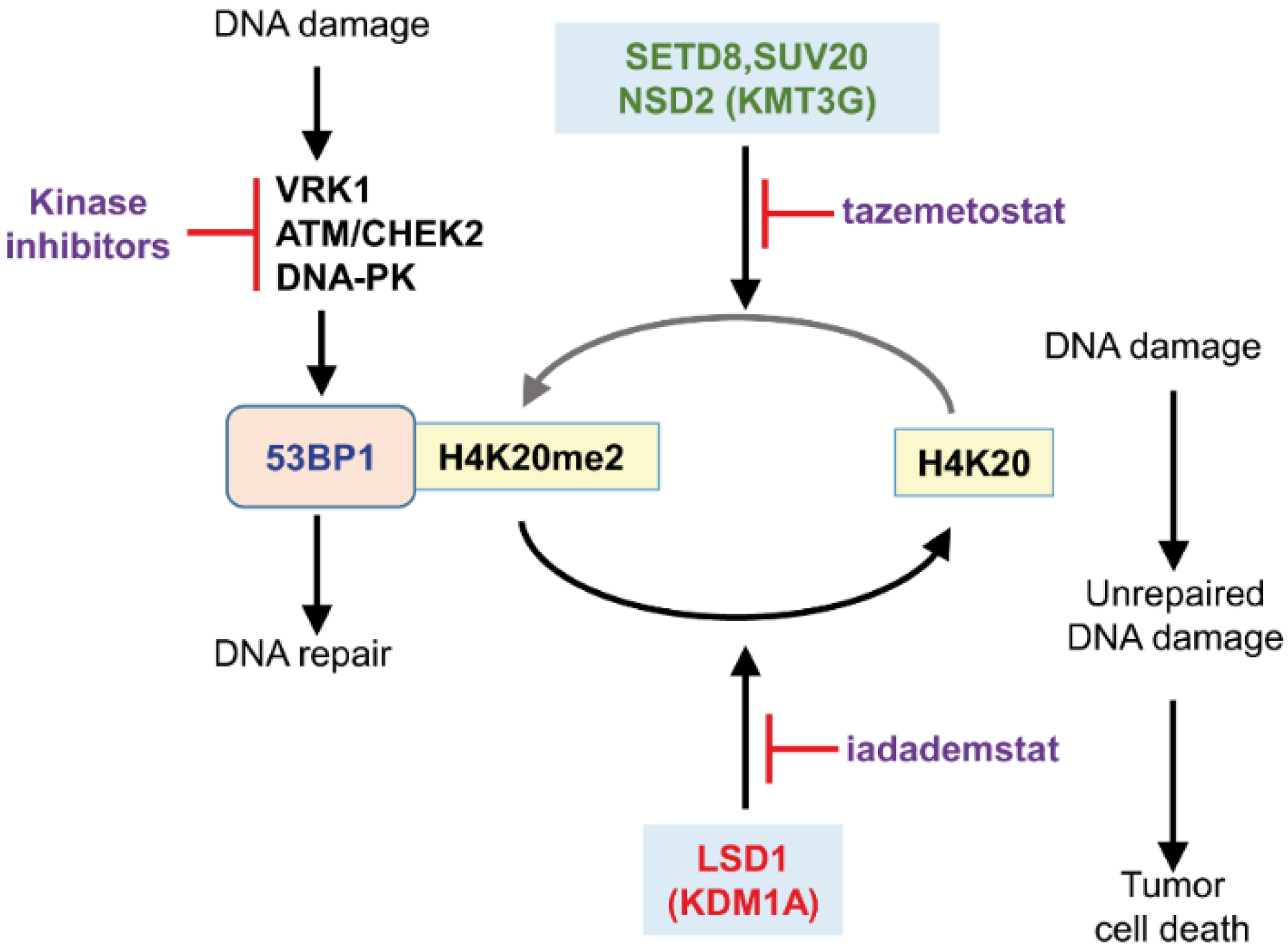
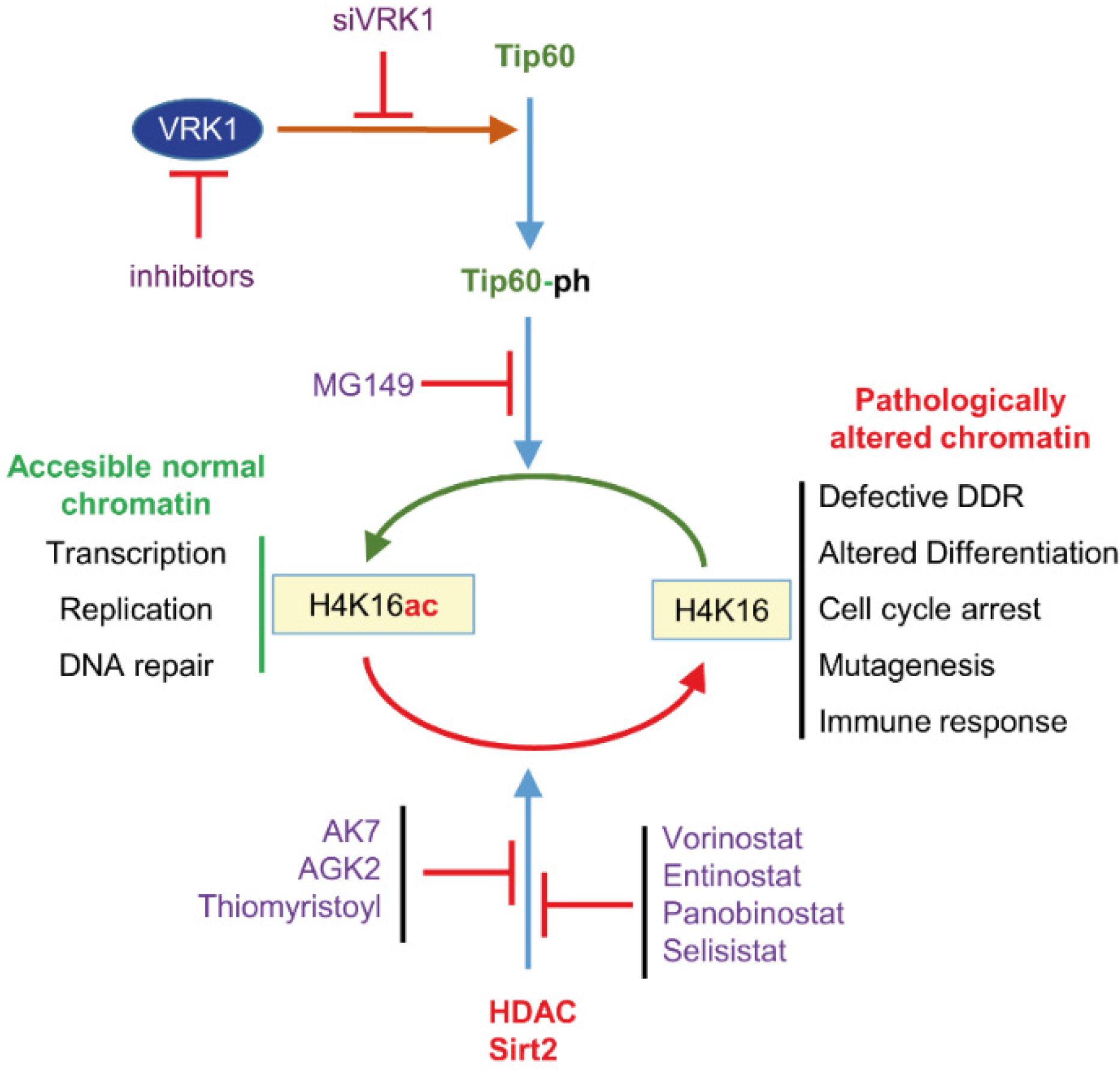
6.2. VRK1
6.3. PI3K (Phosphoinositide 3 Lipid Kinase (PI3K)-Related Protein Kinase) Family
7. Conclusions
Funding
Conflicts of Interest
References
- Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Elias, K.M. Synthetic Lethality in Ovarian Cancer. Mol. Cancer 2021, 20, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Crowley, F.; Park, W.; O’Reilly, E.M. Targeting DNA damage repair pathways in pancreas cancer. Cancer Metastasis Rev. 2021, 40, 891–908. [Google Scholar] [CrossRef]
- Carmichael, J.; Maza, M.; Rescigno, P.; Chandran, K.; de Bono, J. Targeting defective DNA repair in prostate cancer. Curr. Opin. Oncol. 2020, 32, 503–509. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- Li, B.; Li, T.; Pignon, J.C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Campillo-Marcos, I.; García-González, R.; Navarro-Carrasco, E.; Lazo, P.A. The human VRK1 chromatin kinase in cancer biology. Cancer Lett. 2021, 503, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E. Reshaping chromatin after DNA damage: The choreography of histone proteins. J. Mol. Biol. 2015, 427, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [Green Version]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharm. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hashash, A.H.K. Histone H3K27M Mutation in Brain Tumors. Adv. Exp. Med. Biol. 2021, 1283, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Singh, P.K. Histone methyl transferases: A class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur. J. Med. Chem. 2019, 166, 351–368. [Google Scholar] [CrossRef]
- Kari, V.; Raul, S.K.; Henck, J.M.; Kitz, J.; Kramer, F.; Kosinsky, R.L.; Ubelmesser, N.; Mansour, W.Y.; Eggert, J.; Spitzner, M.; et al. The histone methyltransferase DOT1L is required for proper DNA damage response, DNA repair, and modulates chemotherapy responsiveness. Clin. Epigenet. 2019, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharm. Exp. 2014, 350, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eich, M.L.; Athar, M.; Ferguson, J.E., 3rd; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Marcos, I.; Monte-Serrano, E.; Navarro-Carrasco, E.; García-González, R.; Lazo, P.A. Lysine Methyltransferase Inhibitors Impair H4K20me2 and 53BP1 Foci in Response to DNA Damage in Sarcomas, a Synthetic Lethality Strategy. Front. Cell Dev. Biol. 2021, 9, 715126. [Google Scholar] [CrossRef]
- Yuan, H.; Nishikori, M.; Otsuka, Y.; Arima, H.; Kitawaki, T.; Takaori-Kondo, A. The EZH2 inhibitor tazemetostat upregulates the expression of CCL17/TARC in B-cell lymphoma and enhances T-cell recruitment. Cancer Sci. 2021, 112, 4604–4616. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Narendran, A. Targeting EZH2-mediated methylation of histone 3 inhibits proliferation of pediatric acute monocytic leukemia cells in vitro. Cancer Biol. 2021, 22, 333–344. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.-H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e156. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ahn, J.H.; Wang, G.G. Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell Mol. Life Sci. 2019, 76, 2899–2916. [Google Scholar] [CrossRef] [PubMed]
- Coussens, N.P.; Kales, S.C.; Henderson, M.J.; Lee, O.W.; Horiuchi, K.Y.; Wang, Y.; Chen, Q.; Kuznetsova, E.; Wu, J.; Chakka, S.; et al. High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem. 2018, 293, 13750–13765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xie, Y.; Guo, J.; Li, X.; Wang, J.; Jiang, H.; Peng, Z.; Wang, J.; Wang, S.; Li, Q.; et al. Targeting NSD2-mediated SRC-3 liquid-liquid phase separation sensitizes bortezomib treatment in multiple myeloma. Nat. Commun. 2021, 12, 1022. [Google Scholar] [CrossRef]
- Chitale, S.; Richly, H. H4K20me2: Orchestrating the recruitment of DNA repair factors in nucleotide excision repair. Nucleus 2018, 9, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Chitale, S.; Richly, H. DICER- and MMSET-catalyzed H4K20me2 recruits the nucleotide excision repair factor XPA to DNA damage sites. J. Cell Biol. 2018, 217, 527–540. [Google Scholar] [CrossRef]
- Carvalho, S.; Vítor, A.C.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.M.; Ferreira, J.; de Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife 2014, 3, e02482. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, R.; Morejon-Garcia, P.; Campillo-Marcos, I.; Salzano, M.; Lazo, P.A. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers 2020, 12, 2986. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.W.; Alford, J.S.; Boriak-Sjodin, P.A.; Brach, D.; Cosmopoulos, K.; Duncan, K.W.; Eckley, S.T.; Foley, M.A.; Harvey, D.M.; Motwani, V.; et al. Discovery of a First-in-Class Inhibitor of the Histone Methyltransferase SETD2 Suitable for Preclinical Studies. ACS Med. Chem. Lett. 2021, 12, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Kagey, M.H.; Frampton, G.M.; Rahl, P.B.; Young, R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009, 23, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Nakaki, R.; Inagaki, T.; Yoshida, A.; Kano, Y.; Kimura, H.; Tanaka, T.; Tsutsumi, S.; Nakao, M.; Doi, T.; et al. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell 2015, 60, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Paredes, M.; de Paz, A.M.; Simo-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vazquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Flesher, J.L.; Fisher, D.E. G9a: An Emerging Epigenetic Target for Melanoma Therapy. Epigenomes 2021, 5, 23. [Google Scholar] [CrossRef]
- Spiliopoulou, P.; Spear, S.; Mirza, H.; Garner, I.; McGarry, L.; Grundland Freile, F.; Cheng, Z.; Ennis, D.P.; Iyer, N.R.; McNamara, S.; et al. Dual G9A/EZH2 inhibition stimulates anti-tumour immune response in ovarian high grade serous carcinoma. Mol. Cancer Ther. 2022, 21, 522–534. [Google Scholar] [CrossRef]
- Haebe, J.R.; Bergin, C.J.; Sandouka, T.; Benoit, Y.D. Emerging role of G9a in cancer stemness and promises as a therapeutic target. Oncogenesis 2021, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.C.; Burn, T.C.; Sparks, R.; Maduskuie, T.; Diamond, S.; Rupar, M.; Wen, X.; Volgina, A.; Zolotarjova, N.; Waeltz, P.; et al. The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies. Clin. Cancer Res. 2019, 25, 300–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sak, A.; Bannik, K.; Groneberg, M.; Stuschke, M. Chaetocin induced chromatin condensation: Effect on DNA repair signaling and survival. Int. J. Radiat. Biol. 2021, 97, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Seo, I.; Casciello, F.; Jacquelin, S.; Lane, S.W.; Suh, S.I.; Suh, M.H.; Lee, J.S.; Baek, W.K. The anticancer effect of chaetocin is enhanced by inhibition of autophagy. Cell Death Dis. 2016, 7, e2098. [Google Scholar] [CrossRef] [Green Version]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511.e412. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Gumuzio, J.; Verdura, S.; Brunet, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Alarcón, T.; Encinar, J.A.; Martin, Á.G.; Menendez, J.A. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: A potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging 2020, 12, 4794–4814. [Google Scholar] [CrossRef]
- Takagi, S.; Ishikawa, Y.; Mizutani, A.; Iwasaki, S.; Matsumoto, S.; Kamada, Y.; Nomura, T.; Nakamura, K. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res. 2017, 77, 4652–4662. [Google Scholar] [CrossRef] [Green Version]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.P.; McGonagle, A.E.; Wiseman, D.H.; Williams, E.L.; Jordan, A.M. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date. Med. Res. Rev. 2015, 35, 586–618. [Google Scholar] [CrossRef]
- Pedicona, F.; Casado, P.; Hijazi, M.; Gribben, J.G.; Rouault-Pierre, K.; Cutillas, P.R. Targeting the lysine-specific demethylase 1 rewires kinase networks and primes leukemia cells for kinase inhibitor treatment. Sci. Signal. 2022, 15, eabl7989. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544.e10. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef]
- Bose, P.; Konopleva, M.Y. ORY-1001: Overcoming the Differentiation Block in AML. Cancer Cell 2018, 33, 342–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farria, A.; Li, W.; Dent, S.Y. KATs in cancer: Functions and therapies. Oncogene 2015, 34, 4901–4913. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Gupta, V.G.; Liu, Q.; Yull, F.; Crispens, M.A.; Khabele, D. Panobinostat enhances olaparib efficacy by modifying expression of homologous recombination repair and immune transcripts in ovarian cancer. Neoplasia 2022, 24, 63–75. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Laubach, J.P.; Richter, J.; Stricker, S.; Spencer, A.; Richardson, P.G.; Chari, A. Panobinostat From Bench to Bedside: Rethinking the Treatment Paradigm for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk 2021, 21, 752–765. [Google Scholar] [CrossRef]
- Zhang, Y.; Ishida, C.T.; Ishida, W.; Lo, S.L.; Zhao, J.; Shu, C.; Bianchetti, E.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; et al. Combined HDAC and Bromodomain Protein Inhibition Reprograms Tumor Cell Metabolism and Elicits Synthetic Lethality in Glioblastoma. Clin. Cancer Res. 2018, 24, 3941–3954. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; De Carvalho, D.D. Clinical advances in targeting epigenetics for cancer therapy. FEBS J. 2022, 289, 1214–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Tang, X.; Fan, S.; Liu, X.; Sun, J.; Ju, C.; Liang, Y.; Liu, R.; Zhou, R.; Yu, B.; et al. Targeting the p300/NONO axis sensitizes melanoma cells to BRAF inhibitors. Oncogene 2021, 40, 4137–4150. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Rikiishi, H.; Shinohara, F.; Sato, T.; Sato, Y.; Suzuki, M.; Echigo, S. Chemosensitization of oral squamous cell carcinoma cells to cisplatin by histone deacetylase inhibitor, suberoylanilide hydroxamic acid. Int. J. Oncol. 2007, 30, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Xing, H.; Lou, F.; Wang, K.; Pan, Q.; Zhou, X.; Gong, L.; Li, D. Histone deacetylase 2 regulates doxorubicin (Dox) sensitivity of colorectal cancer cells by targeting ABCB1 transcription. Cancer Chemother. Pharm. 2016, 77, 613–621. [Google Scholar] [CrossRef]
- Nagarajan, R.P.; Costello, J.F. Epigenetic mechanisms in glioblastoma multiforme. Semin. Cancer Biol. 2009, 19, 188–197. [Google Scholar] [CrossRef]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenet. 2019, 11, 11. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef] [PubMed]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol. Ther. 2021, 220, 107721. [Google Scholar] [CrossRef] [PubMed]
- Vasilatos, S.N.; Katz, T.A.; Oesterreich, S.; Wan, Y.; Davidson, N.E.; Huang, Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis 2013, 34, 1196–1207. [Google Scholar] [CrossRef]
- Halsall, J.A.; Turan, N.; Wiersma, M.; Turner, B.M. Cells adapt to the epigenomic disruption caused by histone deacetylase inhibitors through a coordinated, chromatin-mediated transcriptional response. Epigenetics Chromatin 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshelava, N.; Davicioni, E.; Wan, Z.; Ji, L.; Sposto, R.; Triche, T.J.; Reynolds, C.P. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J. Natl. Cancer Inst. 2007, 99, 1107–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Syn.nthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Somers, K.; Murray, J.; Pandher, R.; Karsa, M.; Ronca, E.; Bongers, A.; Terry, R.; Ehteda, A.; Gamble, L.D.; et al. Dual Targeting of Chromatin Stability By The Curaxin CBL0137 and Histone Deacetylase Inhibitor Panobinostat Shows Significant Preclinical Efficacy in Neuroblastoma. Clin. Cancer Res. 2021, 27, 4338–4352. [Google Scholar] [CrossRef]
- Park, J.; Thomas, S.; Munster, P.N. Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics 2015, 7, 641–652. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.B.; Kim, C.S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [Green Version]
- Yousafzai, N.A.; Zhou, Q.; Xu, W.; Shi, Q.; Xu, J.; Feng, L.; Chen, H.; Shin, V.Y.; Jin, H.; Wang, X. SIRT1 deacetylated and stabilized XRCC1 to promote chemoresistance in lung cancer. Cell Death Dis. 2019, 10, 363. [Google Scholar] [CrossRef] [Green Version]
- Kalle, A.M.; Mallika, A.; Badiger, J.; Talukdar, P.A.; Sachchidanand. Inhibition of SIRT1 by a small molecule induces apoptosis in breast cancer cells. Biochem. Biophys. Res. Commun. 2010, 401, 13–19. [Google Scholar] [CrossRef]
- Hoffmann, G.; Breitenbucher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Hu, J.; He, B.; Negron Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Brown, J.A.; Bourke, E.; Eriksson, L.A.; Kerin, M.J. Targeting cancer using KAT inhibitors to mimic lethal knockouts. Biochem. Soc. Trans. 2016, 44, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Fisher, J.B.; Koprowski, S.; McAllister, D.; Kim, M.S.; Lough, J. Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev. Dyn. 2009, 238, 2912–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, N.; Izumi, H.; Noguchi, T.; Nakajima, Y.; Ohmiya, Y.; Shiota, M.; Kidani, A.; Tawara, A.; Kohno, K. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J. Biol. Chem. 2008, 283, 18218–18226. [Google Scholar] [CrossRef] [Green Version]
- Van Den Broeck, A.; Nissou, D.; Brambilla, E.; Eymin, B.; Gazzeri, S. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis 2012, 33, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef]
- Ono, H.; Kato, T.; Murase, Y.; Nakamura, Y.; Ishikawa, Y.; Watanabe, S.; Akahoshi, K.; Ogura, T.; Ogawa, K.; Ban, D.; et al. C646 inhibits G2/M cell cycle-related proteins and potentiates anti-tumor effects in pancreatic cancer. Sci. Rep. 2021, 11, 10078. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, A.; Zhai, K.; Huang, Z.; Huang, H.; Zhou, W.; Huang, Q.; Fang, X.; Prager, B.C.; Wang, X.; et al. SATB2 drives glioblastoma growth by recruiting CBP to promote FOXM1 expression in glioma stem cells. EMBO Mol. Med. 2020, 12, e12291. [Google Scholar] [CrossRef]
- Wang, Y.M.; Gu, M.L.; Meng, F.S.; Jiao, W.R.; Zhou, X.X.; Yao, H.P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021, 39, 96–108.e106. [Google Scholar] [CrossRef]
- Abascal, F.; Acosta, R.; Addleman, N.J.; Adrian, J.; Afzal, V.; Aken, B.; Akiyama, J.A.; Jammal, O.A.; Amrhein, H.; Anderson, S.M.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Salzano, M.; Vazquez-Cedeira, M.; Sanz-Garcia, M.; Valbuena, A.; Blanco, S.; Fernandez, I.F.; Lazo, P.A. Vaccinia-related kinase 1 (VRK1) confers resistance to DNA-damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 2014, 5, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Carrasco, E.; Lazo, P.A. VRK1 Depletion Facilitates the Synthetic Lethality of Temozolomide and Olaparib in Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 683038. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Marcos, I.; Lazo, P.A. Olaparib and ionizing radiation trigger a cooperative DNA-damage repair response that is impaired by depletion of the VRK1 chromatin kinase. J. Exp. Clin. Cancer Res. 2019, 38, 203. [Google Scholar] [CrossRef] [Green Version]
- Melms, J.C.; Vallabhaneni, S.; Mills, C.E.; Yapp, C.; Chen, J.Y.; Morelli, E.; Waszyk, P.; Kumar, S.; Deming, D.; Moret, N.; et al. Inhibition of Haspin Kinase Promotes Cell-Intrinsic and Extrinsic Antitumor Activity. Cancer Res. 2020, 80, 798–810. [Google Scholar] [CrossRef]
- Huertas, D.; Soler, M.; Moreto, J.; Villanueva, A.; Martinez, A.; Vidal, A.; Charlton, M.; Moffat, D.; Patel, S.; McDermott, J.; et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene 2012, 31, 1408–1418. [Google Scholar] [CrossRef]
- Gizatullin, F.; Yao, Y.; Kung, V.; Harding, M.W.; Loda, M.; Shapiro, G.I. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006, 66, 7668–7677. [Google Scholar] [CrossRef] [Green Version]
- Hardwicke, M.A.; Oleykowski, C.A.; Plant, R.; Wang, J.; Liao, Q.; Moss, K.; Newlander, K.; Adams, J.L.; Dhanak, D.; Yang, J.; et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol. Cancer 2009, 8, 1808–1817. [Google Scholar] [CrossRef] [Green Version]
- Alcaraz-Sanabria, A.; Nieto-Jiménez, C.; Corrales-Sánchez, V.; Serrano-Oviedo, L.; Andrés-Pretel, F.; Montero, J.C.; Burgos, M.; Llopis, J.; Galán-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction Between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer 2017, 16, 2552–2562. [Google Scholar] [CrossRef] [Green Version]
- Owonikoko, T.K.; Niu, H.; Nackaerts, K.; Csoszi, T.; Ostoros, G.; Mark, Z.; Baik, C.; Joy, A.A.; Chouaid, C.; Jaime, J.C.; et al. Randomized Phase II Study of Paclitaxel plus Alisertib versus Paclitaxel plus Placebo as Second-Line Therapy for SCLC: Primary and Correlative Biomarker Analyses. J. Thorac. Oncol. 2020, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.; Coleman, R.L.; Roszak, A.; Behbakht, K.; Matulonis, U.; Ray-Coquard, I.; Sawrycki, P.; Duska, L.R.; Tew, W.; Ghamande, S.; et al. Alisertib in Combination With Weekly Paclitaxel in Patients With Advanced Breast Cancer or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA Oncol. 2019, 5, e183773. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Stejskal, A.; Cooke, L.S.; Manziello, A.; Morales, C.; Persky, D.O.; Fisher, R.I.; Miller, T.P.; Qi, W. Aurora A inhibitor (MLN8237) plus vincristine plus rituximab is synthetic lethal and a potential curative therapy in aggressive B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2012, 18, 2210–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.I.; Lin, C.P.; Ren, N.; Angus, S.P.; Dittmer, D.P.; Foote, M.; Parton, T.; Bhatt, A.P.; Fedoriw, Y.D.; Roth, D.P.; et al. Inhibition of Aurora A Kinase in Combination with Chemotherapy Induces Synthetic Lethality and Overcomes Chemoresistance in Myc-Overexpressing Lymphoma. Target. Oncol. 2019, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaudon, E.; Nikitorowicz-Buniak, J.; Sourd, L.; Morisset, L.; El Botty, R.; Huguet, L.; Dahmani, A.; Painsec, P.; Nemati, F.; Vacher, S.; et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 2020, 11, 4053. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, Y.; Chen, Y.; Hsieh, J.T.; Kong, Z. The ATM inhibitor KU55933 sensitizes radioresistant bladder cancer cells with DAB2IP gene defect. Int. J. Radiat. Biol. 2015, 91, 368–378. [Google Scholar] [CrossRef]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 2022, 163, 19–26. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Kwon, M.; An, M.; Kim, S.T.; Smith, S.A.; Loembé, A.B.; Mortimer, P.G.S.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2022, 33, 193–203. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smithson, M.; Irwin, R.K.; Williams, G.; McLeod, M.C.; Choi, E.K.; Ganguly, A.; Pepple, A.; Cho, C.S.; Willey, C.D.; Leopold, J.; et al. Inhibition of DNA-PK may improve response to neoadjuvant chemoradiotherapy in rectal cancer. Neoplasia 2022, 25, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.M.; Parry, D.A.; Williams, T.C.; Nadeu, F.; Hindshaw, R.L.; Rios Szwed, D.O.; Nicholson, M.D.; Carroll, P.; Boyle, S.; Royo, R.; et al. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature 2022, 602, 623–631. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Amoussou, N.G.; Bigot, A.; Roussakis, C.; Robert, J.H. Haspin: A promising target for the design of inhibitors as potent anticancer drugs. Drug Discov. Today 2018, 23, 409–415. [Google Scholar] [CrossRef]
- Nishida-Fukuda, H.; Tokuhiro, K.; Ando, Y.; Matsushita, H.; Wada, M.; Tanaka, H. Evaluation of the antiproliferative effects of the HASPIN inhibitor CHR-6494 in breast cancer cell lines. PLoS ONE 2021, 16, e0249912. [Google Scholar] [CrossRef]
- Wang, P.; Hua, X.; Sun, Y.; Li, H.; Bryner, Y.H.; Hsung, R.P.; Dai, J. Loss of haspin suppresses cancer cell proliferation by interfering with cell cycle progression at multiple stages. FASEB J. 2021, 35, e21923. [Google Scholar] [CrossRef]
- Huang, M.; Feng, X.; Su, D.; Wang, G.; Wang, C.; Tang, M.; Paulucci-Holthauzen, A.; Hart, T.; Chen, J. Genome-wide CRISPR screen uncovers a synergistic effect of combining Haspin and Aurora kinase B inhibition. Oncogene 2020, 39, 4312–4322. [Google Scholar] [CrossRef]
- Wang, P.; Hua, X.; Bryner, Y.H.; Liu, S.; Gitter, C.B.; Dai, J. Haspin inhibition delays cell cycle progression through interphase in cancer cells. J. Cell Physiol. 2020, 235, 4508–4519. [Google Scholar] [CrossRef]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2019, 25, 3430–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Lyu, J.; Yang, E.J.; Liu, Y.; Zhang, B.; Shim, J.S. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat. Commun. 2018, 9, 3212. [Google Scholar] [CrossRef] [PubMed]
- Salzano, M.; Sanz-Garcia, M.; Monsalve, D.M.; Moura, D.S.; Lazo, P.A. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015, 10, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Budziszewski, G.R.; Zhao, Y.; Spangler, C.J.; Kedziora, K.M.; Williams, M.R.; Azzam, D.N.; Skrajna, A.; Koyama, Y.; Cesmat, A.P.; Simmons, H.C.; et al. Multivalent DNA and nucleosome acidic patch interactions specify VRK1 mitotic localization and activity. Nucleic Acids Res. 2022, 50, 4355–4371. [Google Scholar] [CrossRef]
- Aihara, H.; Nakagawa, T.; Mizusaki, H.; Yoneda, M.; Kato, M.; Doiguchi, M.; Imamura, Y.; Higashi, M.; Ikura, T.; Hayashi, T.; et al. Histone H2A T120 Phosphorylation Promotes Oncogenic Transformation via Upregulation of Cyclin D1. Mol. Cell 2016, 64, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Campillo-Marcos, I.; Lazo, P.A. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: A therapeutic target? Cell Mol. Life Sci. 2018, 75, 2375–2388. [Google Scholar] [CrossRef] [Green Version]
- Valbuena, A.; Lopez-Sanchez, I.; Lazo, P.A. Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PLoS ONE 2008, 3, e1642. [Google Scholar] [CrossRef]
- Beronja, S.; Janki, P.; Heller, E.; Lien, W.H.; Keyes, B.E.; Oshimori, N.; Fuchs, E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 2013, 501, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Colmenero-Repiso, A.; Gómez-Muñoz, M.A.; Rodríguez-Prieto, I.; Amador-Álvarez, A.; Henrich, K.O.; Pascual-Vaca, D.; Okonechnikov, K.; Rivas, E.; Westermann, F.; Pardal, R.; et al. Identification of VRK1 as a New Neuroblastoma Tumor Progression Marker Regulating Cell Proliferation. Cancers 2020, 12, 3465. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhai, R.; Shen, H.; Song, G.; Wan, F.; Li, Q. VRK1 promotes proliferation, migration, and invasion of gastric carcinoma cells by activating β-catenin. Neoplasma 2021, 68, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Muller, S.; Bullock, A.N.; Schwaller, J.; Sundstrom, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; de Souza Gama, F.H.; Dutra, L.A.; Dos Reis, C.V.; Vasconcelos, S.N.S.; da Silva Santiago, A.; Takarada, J.E.; Di Pillo, F.; Azevedo, H.; Mascarello, A.; et al. Development of Pyridine-based Inhibitors for the Human Vaccinia-related Kinases 1 and 2. ACS Med. Chem. Lett. 2019, 10, 1266–1271. [Google Scholar] [CrossRef]
- So, J.; Mabe, N.W.; Englinger, B.; Moyer, S.M.; Trissal, M.C.; Marques, J.G.; Kwon, J.; Shim, B.; Panditharatna, E.; Jeong, D.; et al. VRK1 is required in VRK2-methylated cancers of the nervous system. bioRxiv 2021. [Google Scholar] [CrossRef]
- Blanco, S.; Klimcakova, L.; Vega, F.M.; Lazo, P.A. The subcellular localization of vaccinia-related kinase-2 (VRK2) isoforms determines their different effect on p53 stability in tumour cell lines. FEBS J. 2006, 273, 2487–2504. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Garcia, M.; Monsalve, D.M.; Sevilla, A.; Lazo, P.A. Vaccinia-related Kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J. Biol. Chem. 2012, 287, 23757–23768. [Google Scholar] [CrossRef] [Green Version]
- Monsalve, D.M.; Campillo-Marcos, I.; Salzano, M.; Sanz-Garcia, M.; Cantarero, L.; Lazo, P.A. VRK1 phosphorylates and protects NBS1 from ubiquitination and proteasomal degradation in response to DNA damage. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 760–769. [Google Scholar] [CrossRef] [Green Version]
- García, M.E.G.; Kirsch, D.G.; Reitman, Z.J. Targeting the ATM Kinase to Enhance the Efficacy of Radiotherapy and Outcomes for Cancer Patients. Semin. Radiat. Oncol. 2022, 32, 3–14. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef] [PubMed]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Baschnagel, A.M.; Elnaggar, J.H.; VanBeek, H.J.; Kromke, A.C.; Skiba, J.H.; Kaushik, S.; Abel, L.; Clark, P.A.; Longhurst, C.A.; Nickel, K.P.; et al. ATR Inhibitor M6620 (VX-970) Enhances the Effect of Radiation in Non-Small Cell Lung Cancer Brain Metastasis Patient-Derived Xenografts. Mol. Cancer 2021, 20, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 2020, 821, 111692. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, X.; Chen, Y.; Cheung, J.C.; Wang, H.; Jiang, J.; de Val, N.; Fox, T.; Gellert, M.; Yang, W. Structure of an activated DNA-PK and its implications for NHEJ. Mol. Cell 2021, 81, 801–810.e803. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Knudsen, K.E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 2022, 82, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Huang, Z.; Zhai, K.; Huang, Q.; Tao, W.; Kim, L.; Wu, Q.; Almasan, A.; Yu, J.S.; Li, X.; et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 2021, 13, abc7275. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [Green Version]




| Targets (HDAC or KAT) | Inhibitor Combinations | Tumor | Ref. | |
|---|---|---|---|---|
| 1 | HDAC | panobinostat | Ovarian cancer cell line | [78] |
| PARP | olaparib | |||
| 2 | HDAC | panobinostat | Multiple myeloma | [79] |
| Proteasome | Bortezomid | |||
| 3 | HDAC | panobinostat | glioblastoma | [80] |
| BRD | OTX015 | |||
| 4 | HDAC | Panobinostat, Pracinostat, Entinostat, Vorinostat, Belinostat | MDS, AML, CML, lymphomas, NSCLC, breast cancer, multiple cancer types | [81] |
| DNMT | Azacitidine/Decitabine | |||
| 5 | Tip60 (KAT5) | MG149 | Lung cane cell lines | [52] |
| DNA damage | doxorubicin | |||
| 6 | P300 (KAT3B) | C646 | Melanoma with BRAF(V600E) | [82] |
| BRAF | Vemurafenib/ AZ628 | |||
| Kinase Targeted | Kinase Inhibition | Drug Combination | Tumor Type | Ref |
|---|---|---|---|---|
| VRK1 | depletion | Doxorubicin, radiation | Lung, sarcoma, glioma and breast cancer cell lines | [122,123,124] |
| depletion | temozolomide | Glioblastoma cell lines | [123] | |
| depletion | Radiation, Olaparib | Glioblastoma cell lines | [123,124] | |
| Haspin | CX-6258 CHR-6494 | Melanoma | [125,126] | |
| Aurora B | VX-680 GSK1070916 | Imatinib resistance | Lung and breast cancer cell lines. | [127,128] |
| Aurora A | alisertib | LY2603618 (CHEK1 inhi.), Paclitaxel | Ovary, breast, SCLC | [129,130,131] |
| Aurora A | MLN8237 | Vincristine + rituximab | Non-Hodgkin lymphoma | [132] |
| MLN8237 | cyclophosphamide | Myc-overeexpressing lymphomas | [133] | |
| PLK1 | volasertib | Breast cancer palciclobib resistance | [134,135] | |
| ATM | KU55933 KU60019 AZ 20 CGK733 | Radiation | Bladder Neuroblastoma | [136] |
| NBS1-ATM | Mirin | Cis-platin | ovarian | [137] |
| ATR | AZ20 ETP-46464 Berzosertib Ceralasertib CGK733 | radiation Olaparib | - Lung cancer Metastatic melanoma, Ovarian resistant to PARP inhibitors | [138,139,140] |
| DNA-PK | Peposertib (M3814) AZD7648 | - Olaparib | Rectal cancer | [141,142] |
| CDK4/6 | trilaciclib | Platinum/etoposide topotecan | ES-SCLC | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazo, P.A. Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer? Cancers 2022, 14, 4050. https://doi.org/10.3390/cancers14164050
Lazo PA. Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer? Cancers. 2022; 14(16):4050. https://doi.org/10.3390/cancers14164050
Chicago/Turabian StyleLazo, Pedro A. 2022. "Targeting Histone Epigenetic Modifications and DNA Damage Responses in Synthetic Lethality Strategies in Cancer?" Cancers 14, no. 16: 4050. https://doi.org/10.3390/cancers14164050