
Improvements in Quality Control and Library Preparation for Targeted Sequencing Allowed Detection of Potentially Pathogenic Alterations in Circulating Cell-Free DNA Derived from Plasma of Brain Tumor Patients

 ,
,  , ,
, ,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
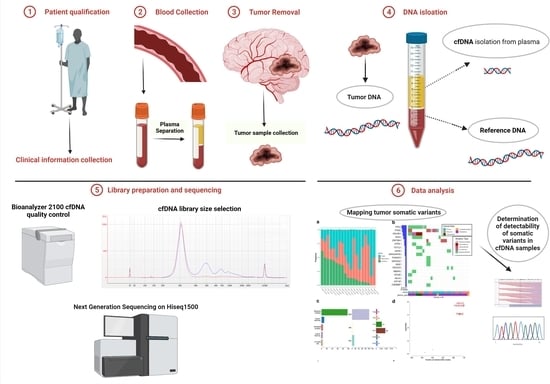
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Isolation
2.3. Design of Targeted Glioma-Related Gene Enrichment Panel
2.4. Sequencing tDNA and gDNA
2.5. ccfDNA Sequencing
2.6. Bioinformatic Analyses
3. Results
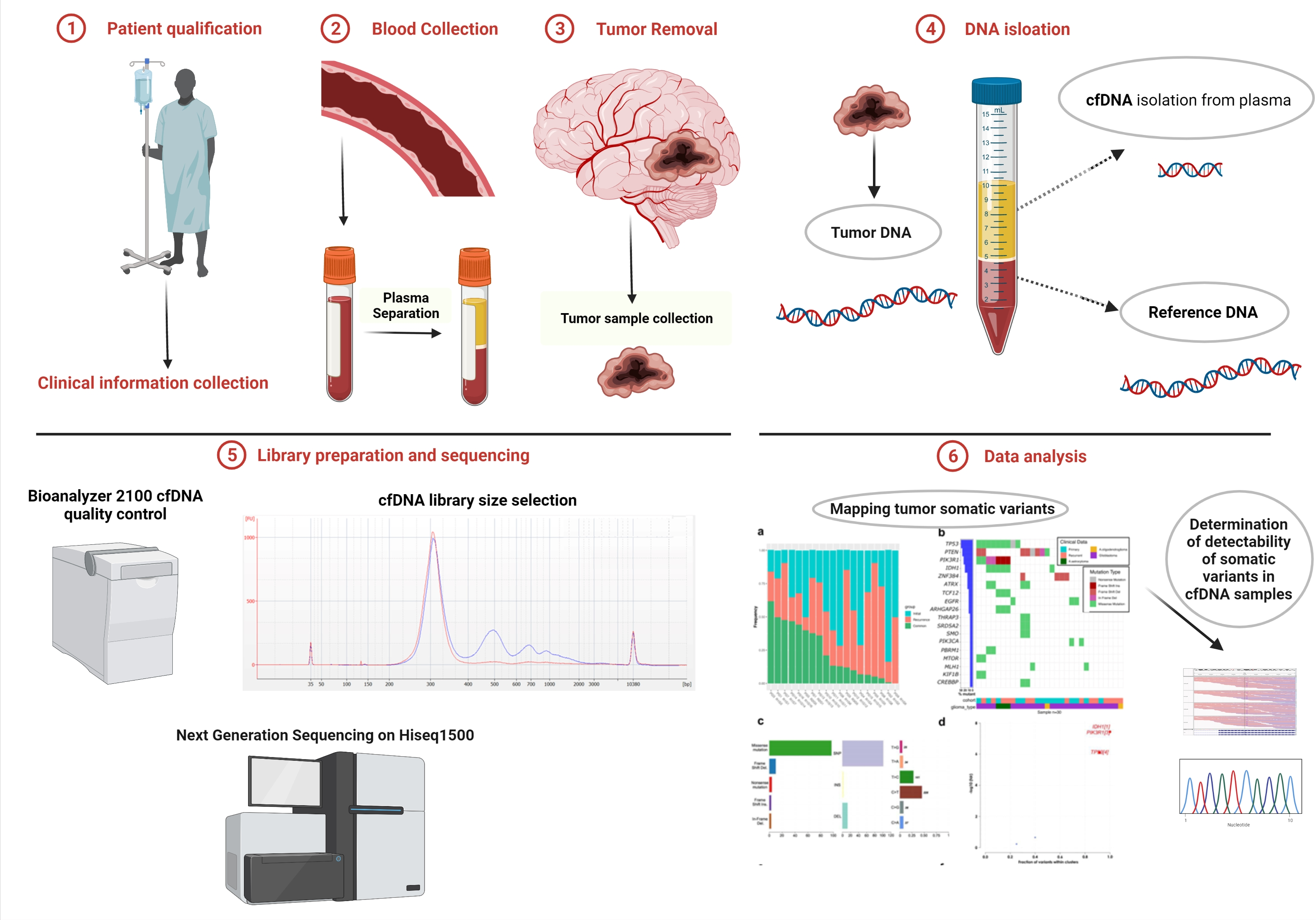
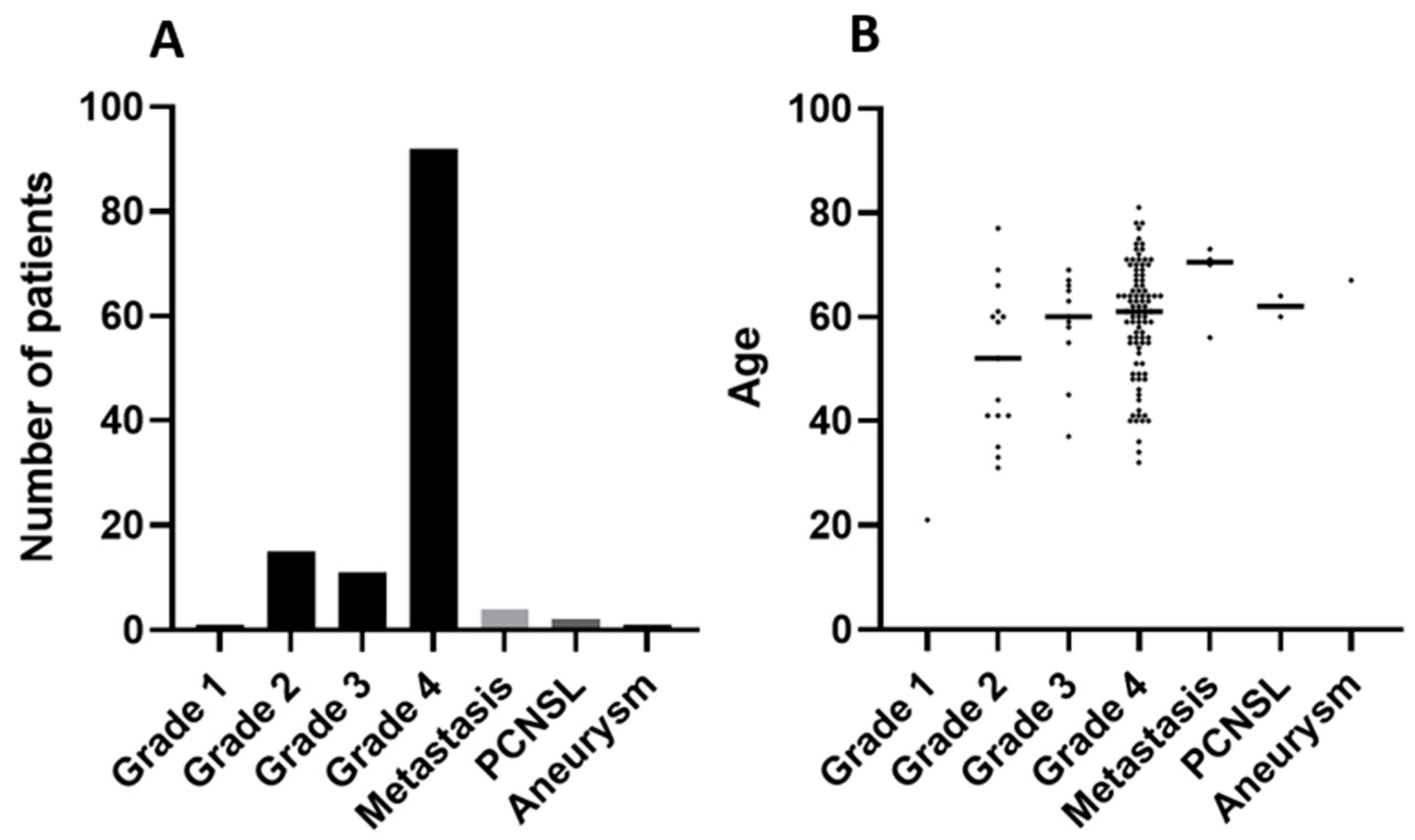
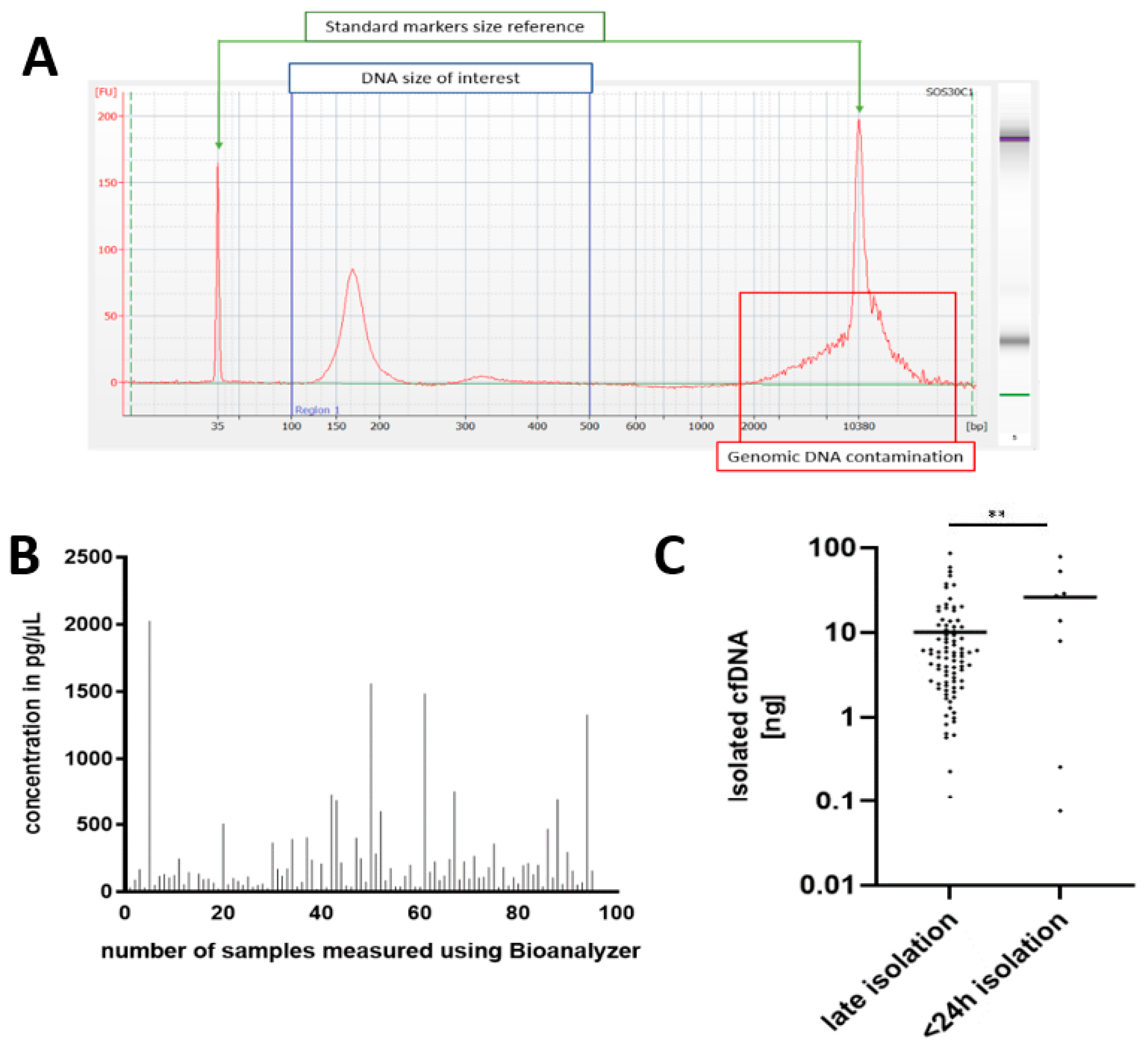
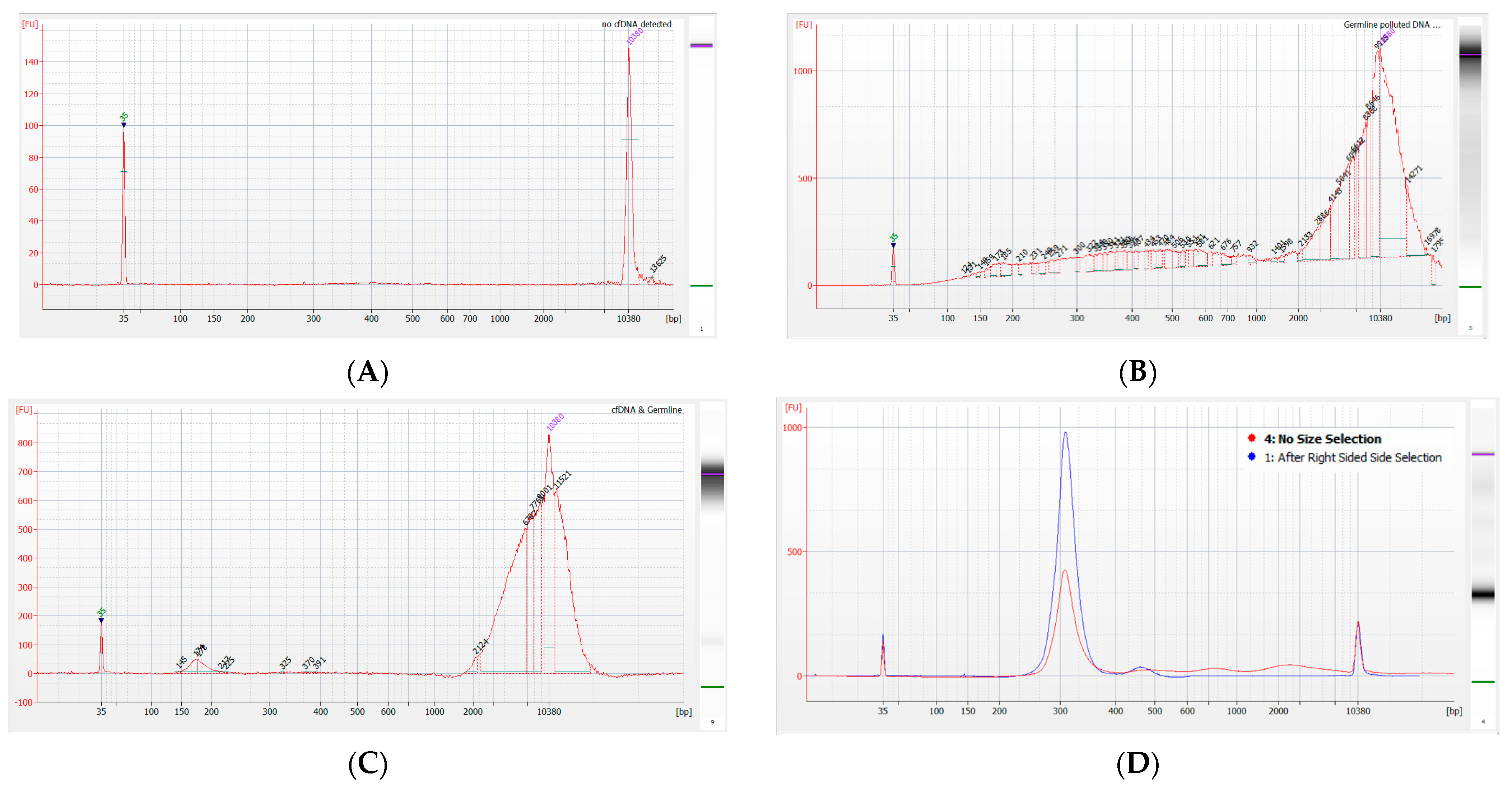
3.1. Cohort Characteristics and Quality Control
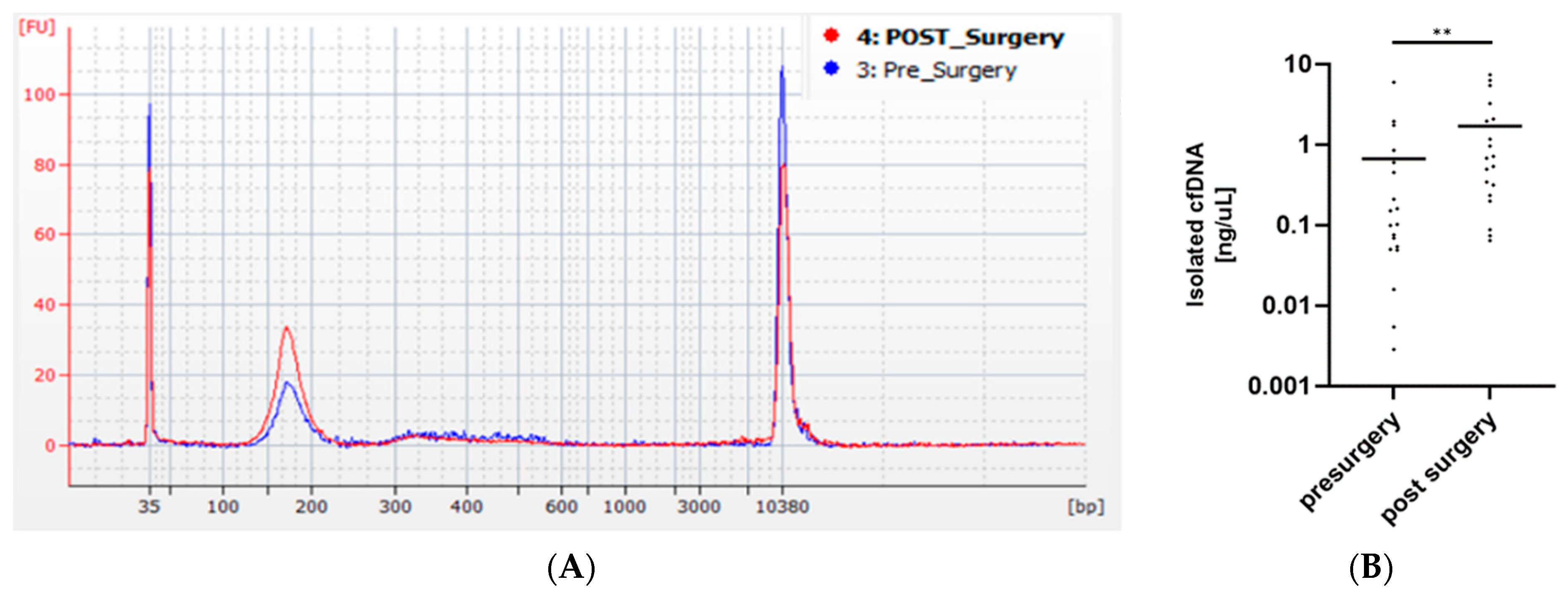
3.2. Precise Quantification of ccfDNA Demonstrates Importance of Fast Blood Processing and Marked Increase in ccfDNA after Surgery
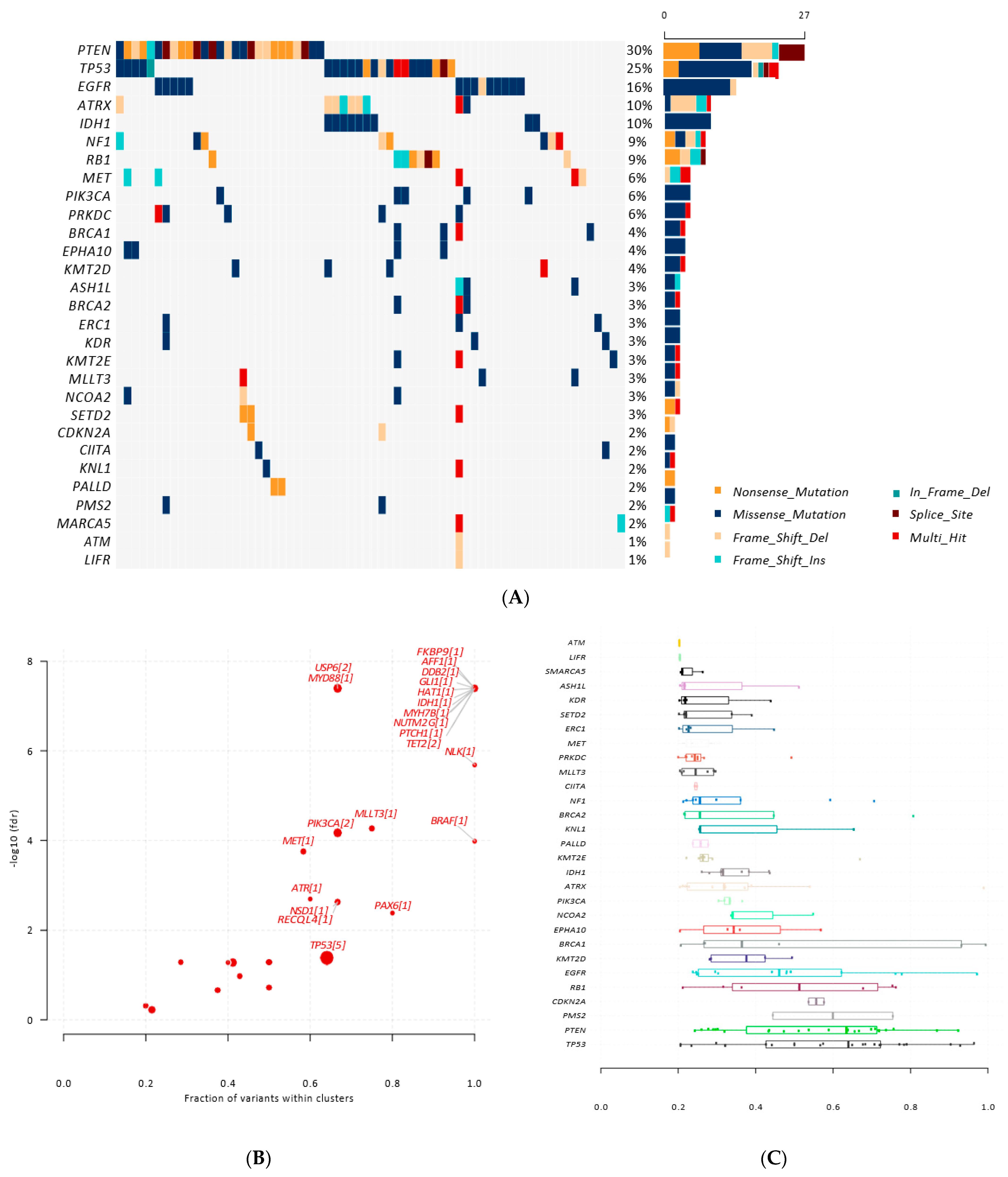
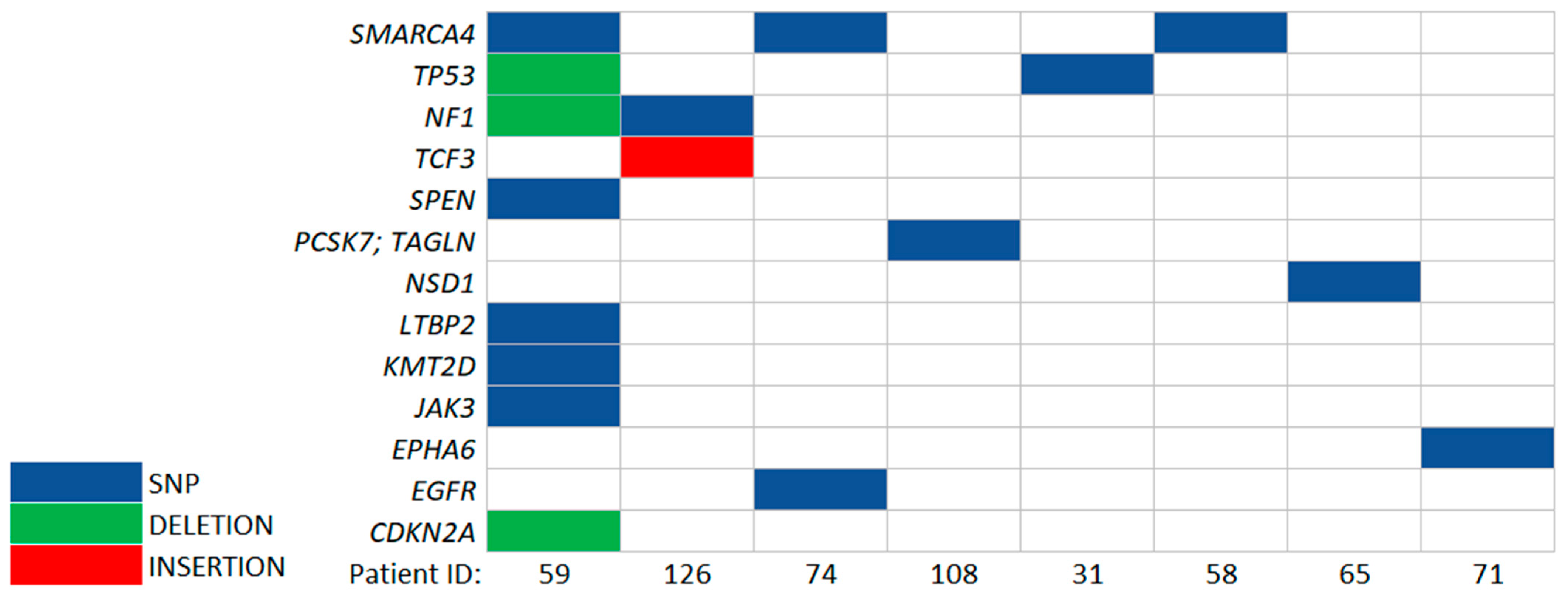
3.3. Identification of Somatic and Germline Variants
3.4. Identification of Somatic Variants in ccfDNA
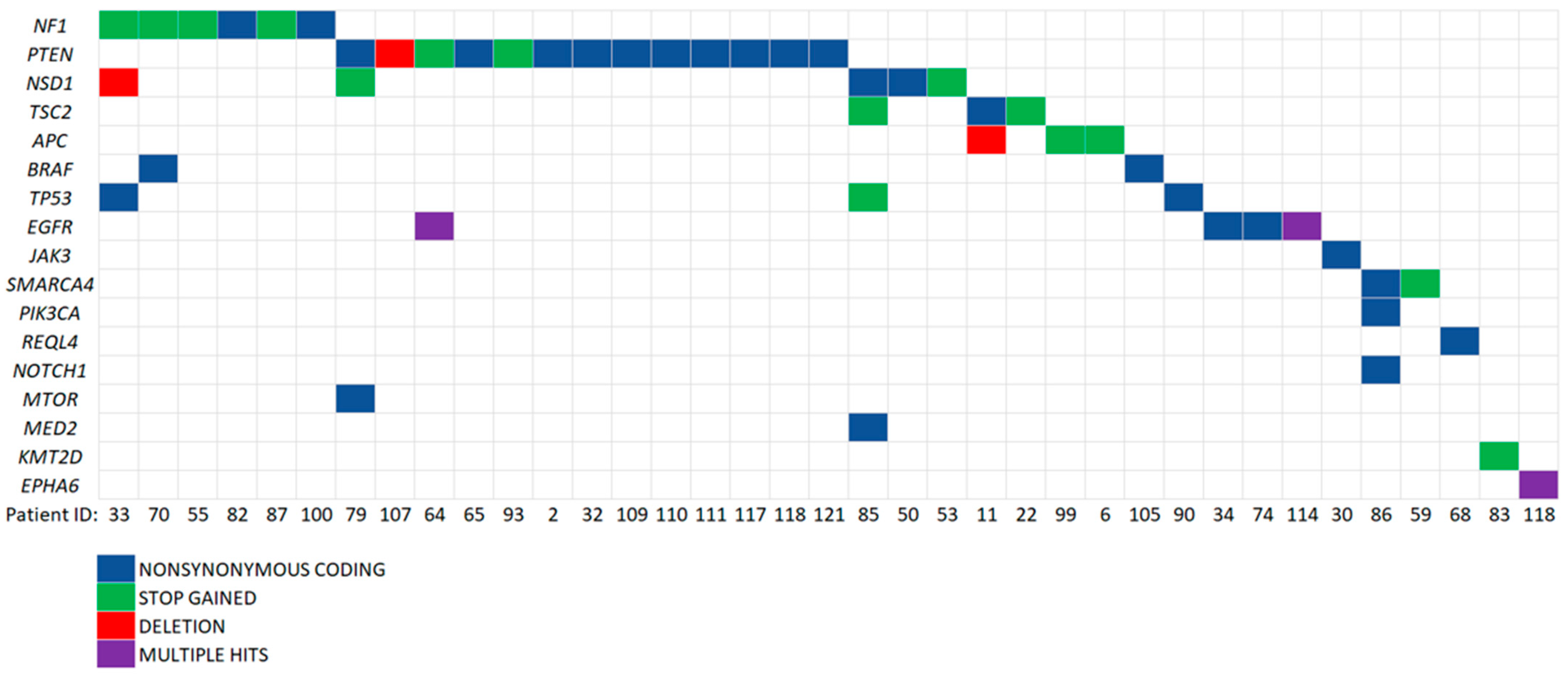
3.5. Potentially Pathogenic Variants Found in ccfDNA, but Not gDNA
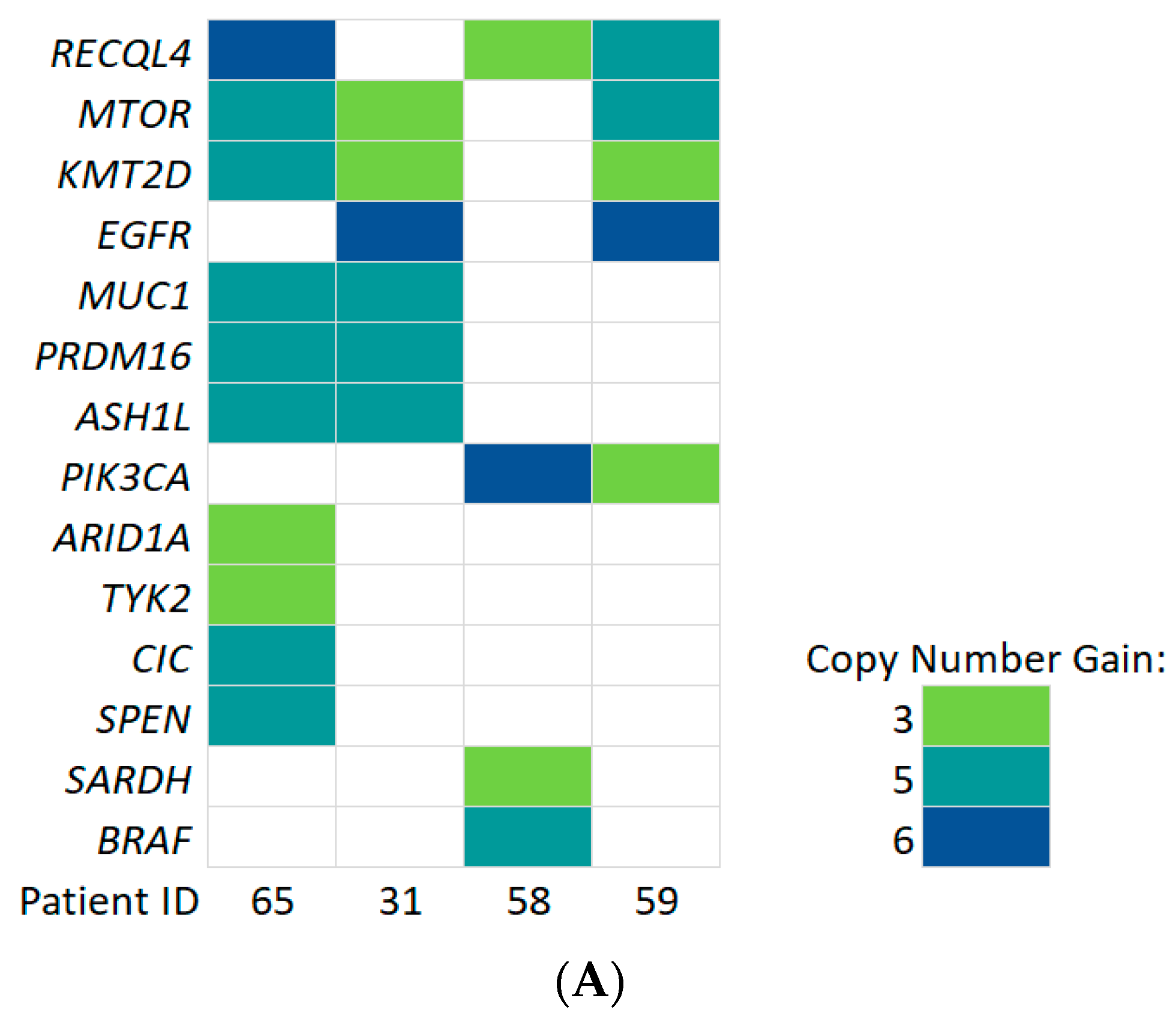
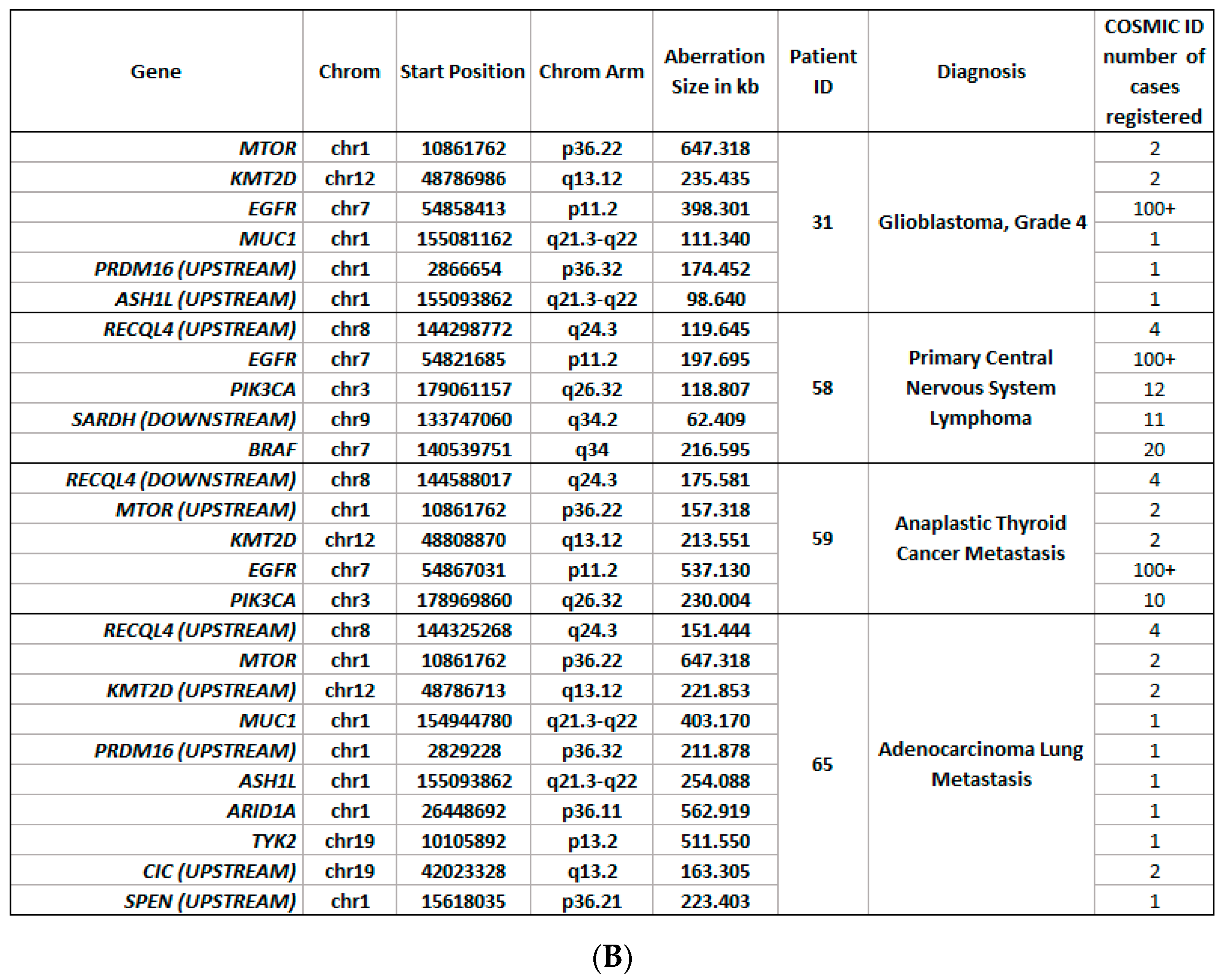
3.6. Detection of Copy Number Alterations in ccfDNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef]
- Jiang, W.-W.; Zahurak, M.; Goldenberg, D.; Milman, Y.; Park, H.L.; Westra, W.H.; Koch, W.; Sidransky, D.; Califano, J. Increased plasma DNA integrity index in head and neck cancer patients. Int. J. Cancer 2006, 119, 2673–2676. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; El Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Chan, C.W.M.; Chan, K.C.A.; Cheng, S.H.; Wong, J.; Wong, V.W.-S.; Wong, G.L.H.; Chan, S.L.; Mok, T.S.K.; Chan, H.L.Y.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, J.; Takinami, Y.; Ueki, M.; Kimura-Kataoka, K.; Yasuda, T.; Takeshita, H. Circulating cell-free DNA fragment analysis by microchip electrophoresis and its relationship with DNase I in cardiac diseases. Clin. Chim. Acta 2019, 497, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Martignetti, J.A.; Camacho-Vanegas, O.; Priedigkeit, N.; Camacho, C.; Pereira, E.; Lin, L.; Garnar-Wortzel, L.; Miller, D.; Losic, B.; Shah, H.; et al. Personalized Ovarian Cancer Disease Surveillance and Detection of Candidate Therapeutic Drug Target in Circulating Tumor DNA. Neoplasia 2014, 16, W28–W29. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Toltl, L.J.; Swystun, L.L.; Pogue, J.; Liaw, K.L.; Weitz, J.I.; Cook, D.J.; Fox-Robichaud, A.E.; Liaw, P.C. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit. Care 2012, 16, R151. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, R.; Li, J.; Zhang, R. Size profile of cell-free DNA: A beacon guiding the practice and innovation of clinical testing. Theranostics 2020, 10, 4737–4748. [Google Scholar] [CrossRef]
- Chan, K.A.; Leung, S.-F.; Yeung, S.-W.; Chan, A.T.; Lo, Y.D. Persistent Aberrations in Circulating DNA Integrity after radiotherapy are associated with poor prognosis in nasopharyngeal carcinoma patients. Clin. Cancer Res. 2008, 14, 4141–4145. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain Tumor Mutations Detected in Cerebral Spinal Fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef]
- Palande, V.; Siegal, T.; Detroja, R.; Gorohovski, A.; Glass, R.; Flueh, C.; Kanner, A.A.; Laviv, Y.; Har-Nof, S.; Levy-Barda, A.; et al. Detection of gene mutations and gene–gene fusions in circulating cell-free DNA of glioblastoma patients: An avenue for clinically relevant diagnostic analysis. Mol. Oncol. 2021, 16, 2098–2114. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 2017, 20, 233–246. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477, Erratum in Cell 2014, 157, 753. [Google Scholar] [CrossRef]
- Kanemaru, Y.; Natsumeda, M.; Okada, M.; Saito, R.; Kobayashi, D.; Eda, T.; Watanabe, J.; Saito, S.; Tsukamoto, Y.; Oishi, M.; et al. Dramatic response of BRAF V600E-mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: Establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol. Commun. 2019, 7, 119. [Google Scholar] [CrossRef]
- Borges, K.S.; Castro-Gamero, A.M.; Moreno, D.A.; Silveira, V.D.S.; Brassesco, M.; Queiroz, R.G.D.P.; de Oliveira, H.F.; Carlotti, C.G.; Scrideli, C.; Tone, L.G. Inhibition of Aurora kinases enhances chemosensitivity to temozolomide and causes radiosensitization in glioblastoma cells. J. Cancer Res. Clin. Oncol. 2011, 138, 405–414. [Google Scholar] [CrossRef]
- Magalhães, T.D.A.; de Sousa, G.R.; Cruzeiro, G.A.V.; Tone, L.G.; Valera, E.T.; Borges, K.S. The therapeutic potential of Aurora kinases targeting in glioblastoma: From preclinical research to translational oncology. Klin. Wochenschr. 2020, 98, 495–512. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Cajal, S.R.Y.; et al. Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumor DNA from cerebrospinal fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Pentsova, E.I.; Shah, R.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J. Clin. Oncol. 2016, 34, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Maleszewska, M.; Kamińska, B. Is glioblastoma an epigenetic malignancy? Cancers 2013, 5, 1120–1139. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Gielniewski, B.; Poleszak, K.; Roura, A.J.; Szadkowska, P.; Krol, S.K.; Guzik, R.; Wiechecka, P.; Maleszewska, M.; Kaza, B.; Marchel, A.; et al. The novel, recurrent mutation in the TOP2A gene results in the enhanced topoisomerase activity and transcription deregulation in glioblastoma. BioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, X.; Lang, J.; Li, S.; Wang, Y.; Peng, L.; Wang, W.; Han, Y.; Qi, C.; Song, L.; Yang, S.; et al. Fragment enrichment of circulating tumor DNA with low-frequency mutations. Front. Genet. 2020, 11, 147. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Von Haeseler, A. NextGenMap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013, 29, 2790–2791. [Google Scholar] [CrossRef]
- Petersen, K.R.; Streett, D.A.; Gerritsen, A.T.; Hunter, S.S.; Settles, M.L. Super deduper, fast PCR duplicate detection in fastq files. In Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics, Atlanta, GA, USA, 9–12 September 2015; pp. 491–492. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Valle, F.D.; Giampieri, E.; Simonetti, G.; Padella, A.; Manfrini, M.; Ferrari, A.; Papayannidis, C.; Zironi, I.; Garonzi, M.; Bernardi, S.; et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinform. 2016, 17, 27–35. [Google Scholar] [CrossRef]
- Roura, A.-J.; Gielniewski, B.; Pilanc, P.; Szadkowska, P.; Maleszewska, M.; Krol, S.K.; Czepko, R.; Kaspera, W.; Wojtas, B.; Kaminska, B. Identification of the immune gene expression signature associated with recurrence of high-grade gliomas. Klin. Wochenschr. 2020, 99, 241–255. [Google Scholar] [CrossRef]
- Arnedo-Pac, C.; Mularoni, L.; Muiños, F.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUSTL: A sequence-based clustering method to identify cancer drivers. Bioinformatics 2019, 35, 4788–4790. [Google Scholar] [CrossRef]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. tumor heterogeneity in glioblastomas: From light microscopy to molecular pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Kim, Y.-W.; Koul, D.; Kim, S.H.; Lucio-Eterovic, A.K.; Freire, P.R.; Yao, J.; Wang, J.; Almeida, J.S.; Aldape, K.; Yung, W.A. Identification of prognostic gene signatures of glioblastoma: A study based on TCGA data analysis. Neuro-Oncology 2013, 15, 829–839. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Bando, H.; Nakamura, Y.; Taniguchi, H.; Shiozawa, M.; Yasui, H.; Esaki, T.; Ohta, T.; Denda, T.; Satoh, T.; Yamazaki, K.; et al. Impact of a metastatic site on circulating tumor DNA (ctDNA) analysis in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, 3554. [Google Scholar] [CrossRef]
- Darrigues, L.; Pierga, J.-Y.; Bernard-Tessier, A.; Bièche, I.; Silveira, A.B.; Michel, M.; Loirat, D.; Cottu, P.; Cabel, L.; Dubot, C.; et al. Circulating tumor DNA as a dynamic biomarker of response to palbociclib and fulvestrant in metastatic breast cancer patients. Breast Cancer Res. 2021, 23, 31. [Google Scholar] [CrossRef]
- Marsavela, G.; McEvoy, A.C.; Pereira, M.R.; Reid, A.L.; Al-Ogaili, Z.; Warburton, L.; Khattak, M.A.; Abed, A.; Meniawy, T.M.; Millward, M.; et al. Detection of clinical progression through plasma ctDNA in metastatic melanoma patients: A comparison to radiological progression. Br. J. Cancer 2021, 126, 401–408. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gDNA | Tumor DNA | ctDNA | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chr | Position | ID | Ref | Alt | Diagnostic Information | Reads | AF | Reads | AF | Reads | AF | |||
| All | Alt | All | Alt | All | Alt | ||||||||||
| TP53 | chr17 | 7577120 | 31 | C | T | Glioblastoma, Grade 4 | 106 | 0 | 0 | 149 | 144 | 0.97 | 1219 | 11 | 0.009 |
| SMARCA4 | chr19 | 11170654 | 58 | G | A | Primary Central Nervous System Lymphoma | 49 | 0 | 0 | 55 | 23 | 0.42 | 602 | 9 | 0.015 |
| SMARCA4 | chr19 | 11144125 | 59 | C | T | Anaplastic Thyroid Cancer Metastasis | 203 | 0 | 0 | 77 | 55 | 0.71 | 1979 | 454 | 0.229 |
| TP53 | chr17 | 7579372 | GC | G | 324 | 0 | 0 | 128 | 76 | 0.59 | 1783 | 435 | 0.244 | ||
| SPEN | chr1 | 16260997 | G | T | 237 | 0 | 0 | 141 | 82 | 0.58 | 2308 | 458 | 0.198 | ||
| KMT2D | chr12 | 49438655 | C | G | 214 | 0 | 0 | 153 | 31 | 0.20 | 2238 | 229 | 0.102 | ||
| LTBP2 | chr14 | 75078119 | T | G | 20 | 0 | 0 | 15 | 9 | 0.60 | 279 | 50 | 0.179 | ||
| NF1 | chr17 | 29560103 | GA | G | 176 | 0 | 0 | 164 | 42 | 0.26 | 2444 | 227 | 0.093 | ||
| CDKN2A | chr9 | 21971193 | GC | G | 148 | 0 | 0 | 95 | 66 | 0.69 | 1199 | 180 | 0.150 | ||
| JAK3 | chr19 | 17952151 | G | T | 29 | 0 | 0 | 9 | 5 | 0.56 | 1246 | 270 | 0.217 | ||
| NSD1 | chr5 | 176720936 | 65 | G | C | Adenocarcinoma Lung Metastasis | 390 | 0 | 0 | 418 | 191 | 0.46 | 862 | 183 | 0.212 |
| EPHA6 | chr3 | 96728829 | 71 | G | GTT | Glioblastoma, Grade 4 | 11 | 0 | 0 | 23 | 3 | 0.13 | 618 | 14 | 0.023 |
| SMARCA4 | chr19 | 11144182 | 74 | G | A | Astrocytoma Anaplasticum, Grade 3 | 43 | 0 | 0 | 259 | 76 | 0.29 | 1602 | 17 | 0.011 |
| EGFR | chr7 | 55210075 | T | G | 123 | 0 | 0 | 3020 | 1514 | 0.50 | 1694 | 427 | 0.252 | ||
| PCSK7;TAGLN | chr11 | 117076708 | 108 | T | C | Glioblastoma, Grade 4 | 12 | 0 | 0 | 67 | 10 | 0.15 | 1539 | 340 | 0.221 |
| NF1 | chr17 | 29563087 | 126 | T | G | Glioblastoma, Grade 4 | 67 | 0 | 0 | 112 | 3 | 0.03 | 2141 | 36 | 0.017 |
| TCF3 | chr19 | 1619749 | A | AGGGTG | 38 | 0 | 0 | 73 | 15 | 0.21 | 1281 | 310 | 0.242 | ||
| gDNA (Maftools) | ccfDNA (SureCall) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chrom | Position | ID | rs ID | MAF | AF 1000 G | gnomAD | ClinVar clinsig | Diagnostic Information | Reads | AF | Reads | AF | ||
| All | Alt | All | Alt | ||||||||||||
| APC | chr5 | 112177901 | 6 | rs752654519 | - | - | - | pathogenic/likely pathogenic | Glioblastoma, Grade 4 | 225 | 0 | 0 | 201 | 5 | 0.0249 |
| TSC2 | chr16 | 2098642 | 11 | rs397515228 | - | - | - | pathogenic | Diffuse Glioma, Grade 2 | 204 | 0 | 0 | 303 | 6 | 0.0198 |
| APC | chr5 | 112111411 | rs886039642 | - | - | - | pathogenic/likely pathogenic | 168 | 0 | 0 | 172 | 4 | 0.0233 | ||
| TSC2 | chr16 | 2136203 | 22 | rs45517360 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 53 | 0 | 0 | 101 | 6 | 0.0594 |
| JAK3 | chr19 | 17950375 | 30 | rs145751599 | 0 | 0.0004 | 2 × 10−5 | uncertain significance | Glioblastoma, Grade 4 | 212 | 0 | 0 | 200 | 4 | 0.02 |
| NF1 | chr17 | 29677233 | 31 | rs377662483 | 0 | 0.0002 | 2 × 10−5 | uncertain significance | Glioblastoma, Grade 4 | 129 | 0 | 0 | 814 | 9 | 0.0111 |
| NF1 | chr17 | 29654553 | 33 | rs876657714 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 252 | 0 | 0 | 470 | 4 | 0.00851 |
| TP53 | chr17 | 7577586 | rs587781589 | - | - | - | pathogenic | 239 | 0 | 0 | 345 | 3 | 0.0087 | ||
| NSD1 | chr5 | 176637449 | rs587784080 | - | - | - | pathogenic | 250 | 0 | 0 | 557 | 5 | 0.00898 | ||
| EGFR | chr7 | 55233043 | 34 | rs139236063 | - | - | 4 × 10−6 | likely pathogenic | Glioblastoma, Grade 4 | 120 | 0 | 0 | 2897 | 39 | 0.0135 |
| NSD1 | chr5 | 176709524 | 50 | rs587784169 | - | - | - | pathogenic | Diffuse Astrocytoma, Grade 2 | 154 | 0 | 0 | 428 | 4 | 0.00935 |
| NSD1 | chr5 | 176696631 | 53 | rs794727176 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 239 | 0 | 0 | 335 | 3 | 0.00896 |
| NF1 | chr17 | 29486070 | 55 | rs746824139 | - | - | 0 | pathogenic | Glioblastoma, Grade 4 | 144 | 0 | 0 | 424 | 5 | 0.0118 |
| PTEN | chr10 | 89717695 | 64 | rs190070312 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 246 | 0 | 0 | 436 | 5 | 0.0115 |
| PTEN | chr10 | 89711900 | 65 | rs121913294 | - | - | - | likely pathogenic | Adenocarcinoma Lung Metastasis | 139 | 0 | 0 | 352 | 3 | 0.00852 |
| RECQL4 | chr8 | 145741409 | 68 | rs549497811 | 0 | 0.0002 | 2 × 10−5 | uncertain significance | Glioblastoma, Grade 4 | 240 | 0 | 0 | 563 | 9 | 0.016 |
| BRAF | chr7 | 140454008 | 70 | rs397516894 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 228 | 0 | 0 | 372 | 4 | 0.0108 |
| NF1 | chr17 | 29562981 | rs376576925 | - | - | 4 × 10−6 | pathogenic | 195 | 0 | 0 | 613 | 6 | 0.00979 | ||
| NF1 | chr17 | 29560088 | rs878853884 | - | - | - | pathogenic | 118 | 0 | 0 | 626 | 8 | 0.0128 | ||
| MTOR | chr1 | 11184573 | 79 | rs587777894 | - | - | - | pathogenic | Glioblastoma with Oligodendroglioma Component, Grade 4 | 133 | 0 | 0 | 250 | 8 | 0.032 |
| NSD1 | chr5 | 176673711 | rs570278338 | - | - | - | pathogenic | 65 | 0 | 0 | 132 | 2 | 0.0152 | ||
| PTEN | chr10 | 89692793 | rs786204927 | - | - | - | likely pathogenic | 99 | 0 | 0 | 180 | 7 | 0.0389 | ||
| NF1 | chr17 | 29677228 | 82 | rs533110479 | 0 | 0.0002 | 3 × 10−5 | uncertain significance | Glioblastoma, Grade 4 | 243 | 0 | 0 | 436 | 5 | 0.0115 |
| KMT2D | chr12 | 49438067 | 83 | rs886043414 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 144 | 0 | 0 | 143 | 2 | 0.014 |
| TP53 | chr17 | 7579529 | 85 | rs876658483 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 198 | 0 | 0 | 347 | 4 | 0.0115 |
| TSC2 | chr16 | 2114342 | rs45517179 | - | - | - | pathogenic | 248 | 0 | 0 | 393 | 4 | 0.0102 | ||
| MED12 | chrX | 70357138 | rs762659794 | 0 | 0.0003 | 6 × 10−6 | uncertain significance | 115 | 0 | 0 | 161 | 4 | 0.0248 | ||
| PIK3CA | chr3 | 178952085 | 86 | rs121913279 | - | - | 4 × 10−6 | pathogenic FDA recognized | Giant Cell Glioblastoma, Grade 4 | 245 | 0 | 0 | 139 | 2 | 0.0144 |
| NOTCH1 | chr9 | 139395108 | rs371414501 | 0 | 0.0002 | 2 × 10−5 | uncertain significance | 178 | 0 | 0 | 165 | 3 | 0.0182 | ||
| SMARCA4 | chr19 | 11094931 | rs563079629 | 0 | 0.0002 | 5 × 10−5 | uncertain significance | 58 | 0 | 0 | 136 | 5 | 0.0368 | ||
| NF1 | chr17 | 29588751 | 87 | rs760703505 | - | - | 8 × 10−6 | pathogenic/likely pathogenic | Glioblastoma, Grade 4 | 241 | 0 | 0 | 485 | 4 | 0.00825 |
| PTEN | chr10 | 89720768 | 93 | rs746930141 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 70 | 0 | 0 | 199 | 2 | 0.0101 |
| APC | chr5 | 112173704 | 99 | rs587779783 | - | - | - | pathogenic | Diffuse Astrocytoma, Grade 2 | 250 | 0 | 0 | 643 | 6 | 0.00933 |
| NF1 | chr17 | 29490394 | 100 | rs199474752 | - | - | - | likely pathogenic | Glioblastoma, Grade 4 | 168 | 0 | 0 | 356 | 3 | 0.00843 |
| BRAF | chr7 | 140453137 | 105 | rs121913378 | - | - | - | likely pathogenic | Pleomorphic Xanthoastrocytoma, Grade 2 | 209 | 0 | 0 | 273 | 8 | 0.0293 |
| PTEN | chr10 | 89711968 | 107 | rs587776670 | - | - | - | pathogenic | Glioblastoma, Grade 4 | 104 | 0 | 0 | 482 | 4 | 0.0083 |
| gDNA | ccfDNA | Tumor DNA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | ID | rs ID | COSMIC (CNS)/Polyphen Pred | GMAF | ClinVar Clinsig | Diagnostic Information | Reads | AF | Reads | AF | AF (Somatic in Other Patient) | ||
| All | Alt | All | Alt | ||||||||||
| PTEN | 2 | rs12573787 | -/- | 0.16 | benign | Oligoastrocytoma, Grade 2 | 60 | 0 | 0.00 | 255 | 8 | 0.031 | 0.6923 |
| 32 | Oligodendroglioma Anaplasticum, Grade 3 | 79 | 1 | 0.01 | 150 | 62 | 0.413 | ||||||
| 65 | Adenocarcinoma Lung Metastasis | 55 | 1 | 0.02 | 61 | 33 | 0.541 | ||||||
| 85 | Glioblastoma, Grade 4 | 56 | 1 | 0.02 | 260 | 140 | 0.538 | ||||||
| 109 | Anaplastic Pleomorphic Xantoastrocytoma, Grade 3 | 57 | 0 | 0.00 | 168 | 13 | 0.077 | ||||||
| 110 | Glioblastoma, Grade 4 | 39 | 2 | 0.05 | 234 | 144 | 0.615 | ||||||
| 111 | Glioblastoma, Grade 4 | 107 | 1 | 0.01 | 160 | 68 | 0.425 | ||||||
| 117 | Glioblastoma, Grade 4 | 73 | 1 | 0.01 | 299 | 144 | 0.482 | ||||||
| 118 | Glioblastoma, Grade 4 | 40 | 1 | 0.03 | 224 | 114 | 0.509 | ||||||
| 121 | Giant Cell Glioblastoma, Grade 4 | 52 | 0 | 0.00 | 340 | 164 | 0.482 | ||||||
| TP53 | 90 | rs121913343 | 131/D | - | pathogenic/likely pathogenic | Glioblastoma, Grade 4 | 147 | 3 | 0.02 | 1313 | 27 | 0.021 | 0.2619 |
| EGFR | 64 | rs1057519828 | 14/D | - | likely pathogenic | Glioblastoma, Grade 4 | 225 | 0 | 0.00 | 612 | 22 | 0.036 | 0.4502 |
| EGFR | 64 | rs149840192 | 36/D | - | likely pathogenic | Glioblastoma, Grade 4 | 181 | 3 | 0.02 | 703 | 9 | 0.013 | 0.248 |
| gDNA (Maftools) | ccfDNA (SureCall) | Tumor | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chr | Position | Ref | Alt | ID | rs ID | Registered in COSMIC | ClinVar Clinsig | Diagnostic Information | Reads | AF | Reads | AF | # of Patients with Somatic Variant | ||
| All | Alt | All | Alt | |||||||||||||
| SMARCA4 | 19 | 11144125 | C | T | 59 | yes | - | Anaplastic Thyroid Cancer Metastasis | 186 | 0 | 0 | 1979 | 454 | 0.229409 | 1 | |
| PIK3CA | 3 | 178952085 | A | G | 86 | rs121913279 | - | likely pathogenic | Giant Cell Glioblastoma, Grade 4 | 245 | 0 | 0 | 139 | 2 | 0.014388 | 1 |
| EPHA6 | 3 | 97365038 | G | A | 118 | rs301948 | - | - | Glioblastoma, Grade 4 | 199 | 0 | 0 | 1328 | 86 | 0.064759 | 1 |
| EPHA6 | 3 | 97365074 | A | G | 118 | rs301949 | yes | - | Glioblastoma, Grade 4 | 179 | 0 | 0 | 1611 | 103 | 0.063935 | 1 |
| EGFR | 7 | 55210075 | T | G | 74 | yes | - | Astrocytoma Anaplasticum, Grade 3 | 229 | 0 | 0 | 1694 | 427 | 0.252066 | 2 | |
| EGFR | 7 | 55210075 | T | G | 114 | yes | - | Astrocytoma, Grade 3 | 247 | 1 | 0.004 | 274 | 3 | 0.010949 | 2 | |
| EGFR | 7 | 55224307 | C | T | 114 | yes | likely pathogenic | Astrocytoma, Grade 3 | 245 | 0 | 0 | 417 | 9 | 0.021583 | 1 | |
| EGFR | 7 | 55221822 | C | T | 64 | rs149840192 | yes | - | Glioblastoma, Grade 4 | 181 | 3 | 0.0166 | 703 | 9 | 0.012802 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szadkowska, P.; Roura, A.-J.; Wojtas, B.; Wojnicki, K.; Licholai, S.; Waller, T.; Gubala, T.; Zukowski, K.; Karpeta, M.; Wilkus, K.; et al. Improvements in Quality Control and Library Preparation for Targeted Sequencing Allowed Detection of Potentially Pathogenic Alterations in Circulating Cell-Free DNA Derived from Plasma of Brain Tumor Patients. Cancers 2022, 14, 3902. https://doi.org/10.3390/cancers14163902
Szadkowska P, Roura A-J, Wojtas B, Wojnicki K, Licholai S, Waller T, Gubala T, Zukowski K, Karpeta M, Wilkus K, et al. Improvements in Quality Control and Library Preparation for Targeted Sequencing Allowed Detection of Potentially Pathogenic Alterations in Circulating Cell-Free DNA Derived from Plasma of Brain Tumor Patients. Cancers. 2022; 14(16):3902. https://doi.org/10.3390/cancers14163902
Chicago/Turabian StyleSzadkowska, Paulina, Adria-Jaume Roura, Bartosz Wojtas, Kamil Wojnicki, Sabina Licholai, Tomasz Waller, Tomasz Gubala, Kacper Zukowski, Michal Karpeta, Kinga Wilkus, and et al. 2022. "Improvements in Quality Control and Library Preparation for Targeted Sequencing Allowed Detection of Potentially Pathogenic Alterations in Circulating Cell-Free DNA Derived from Plasma of Brain Tumor Patients" Cancers 14, no. 16: 3902. https://doi.org/10.3390/cancers14163902