
A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications

 ,
,  ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
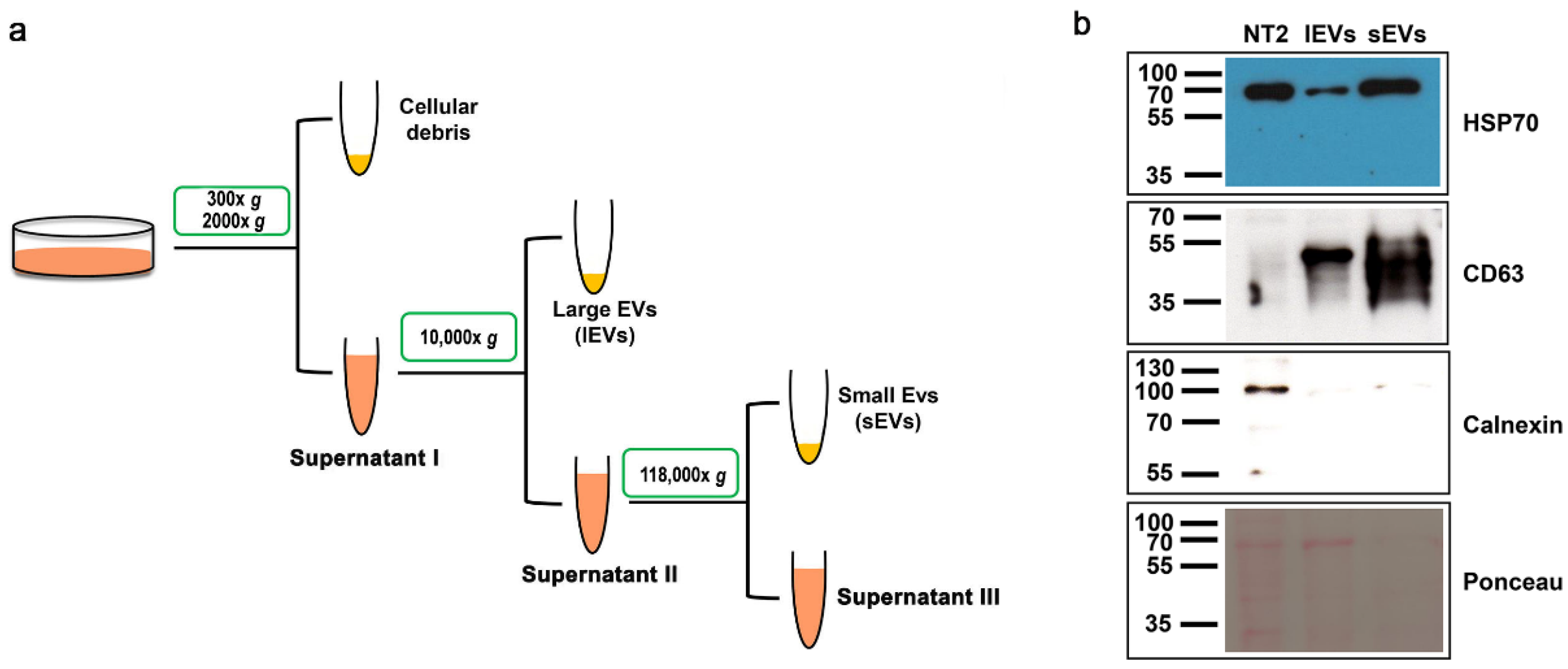
2.2. Extracellular Vesicle Isolation
2.3. Cell Lysate Preparation
2.4. Western Blot
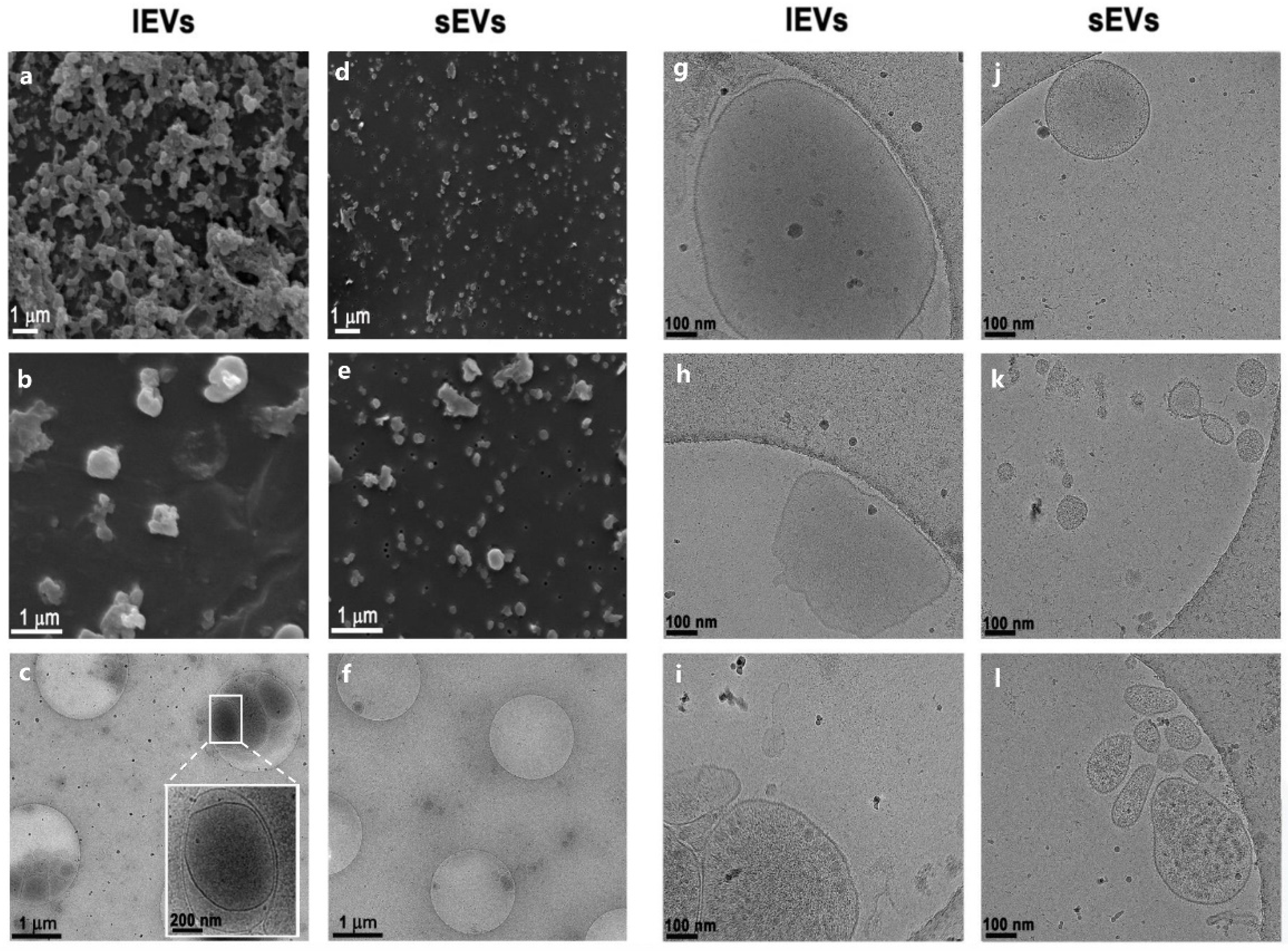
2.5. Scanning Electron Microscopy
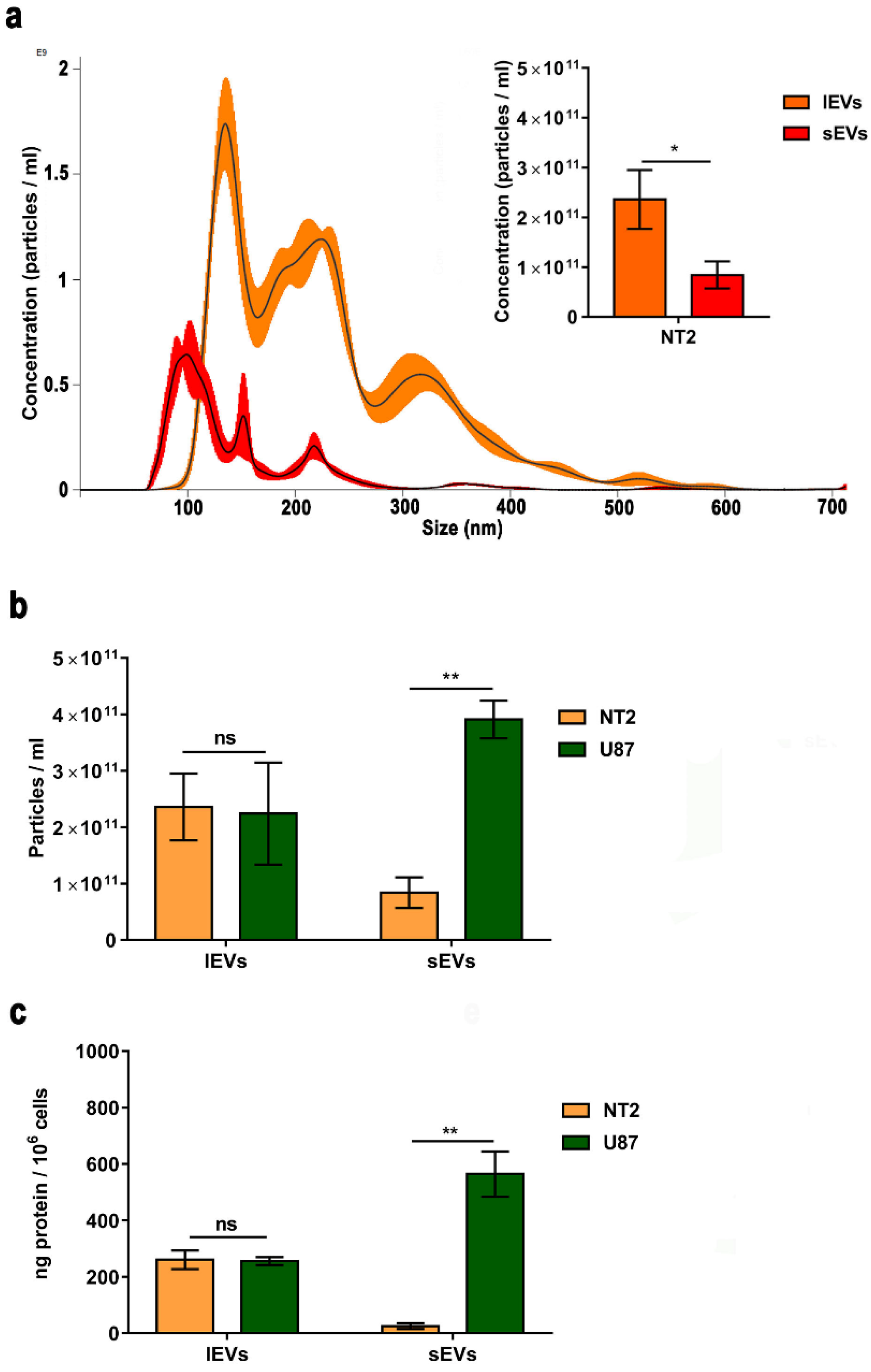
2.6. Nanoparticle Tracking Analysis
2.7. Cryogenic Transmission Electron Microscopy
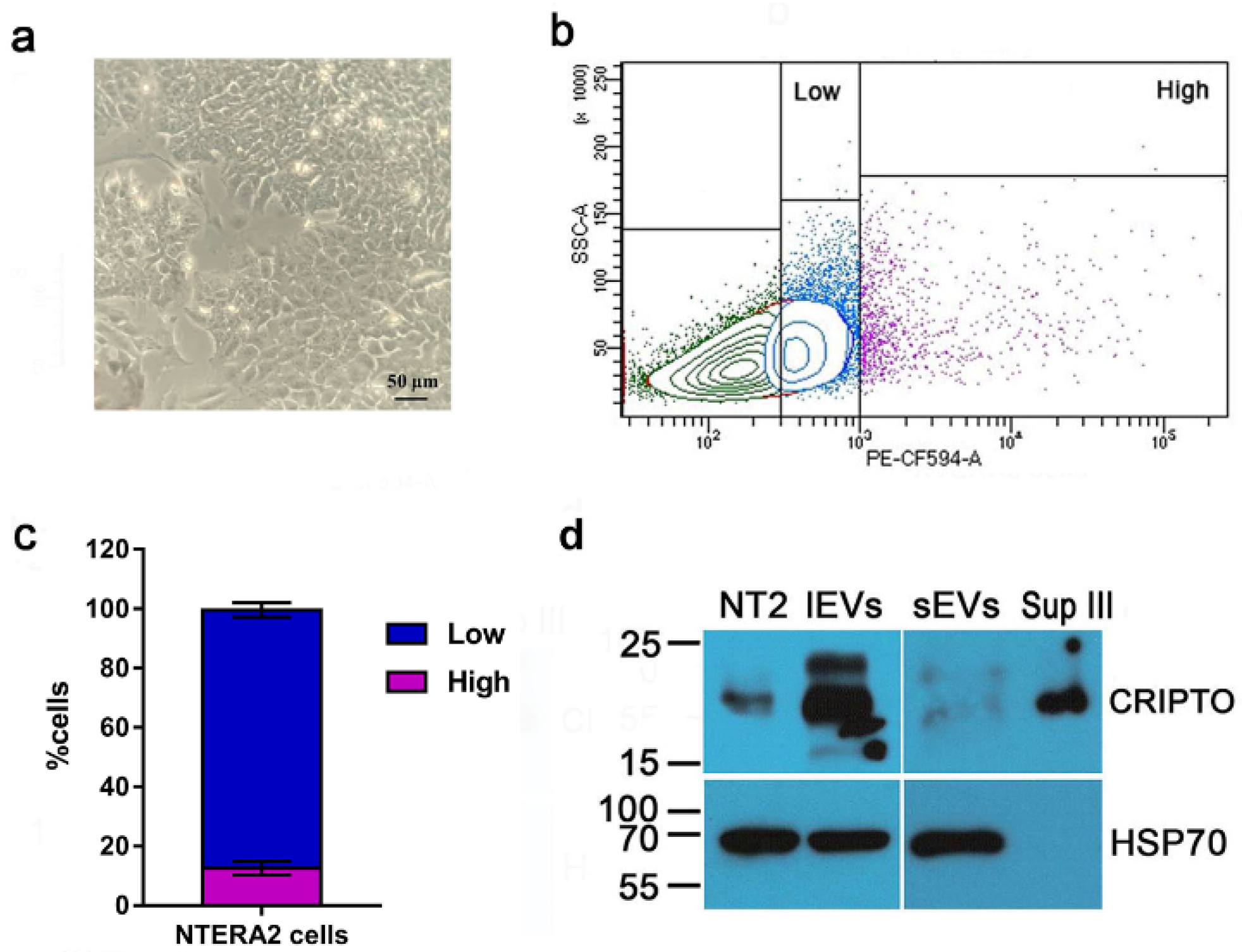
2.8. Flow Cytometry
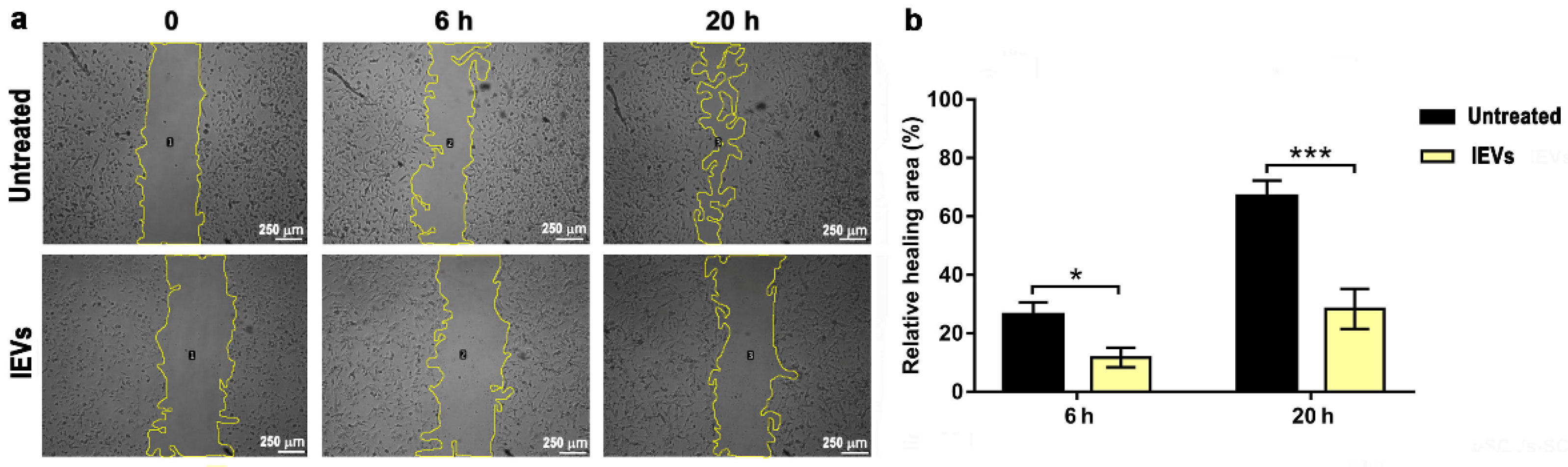
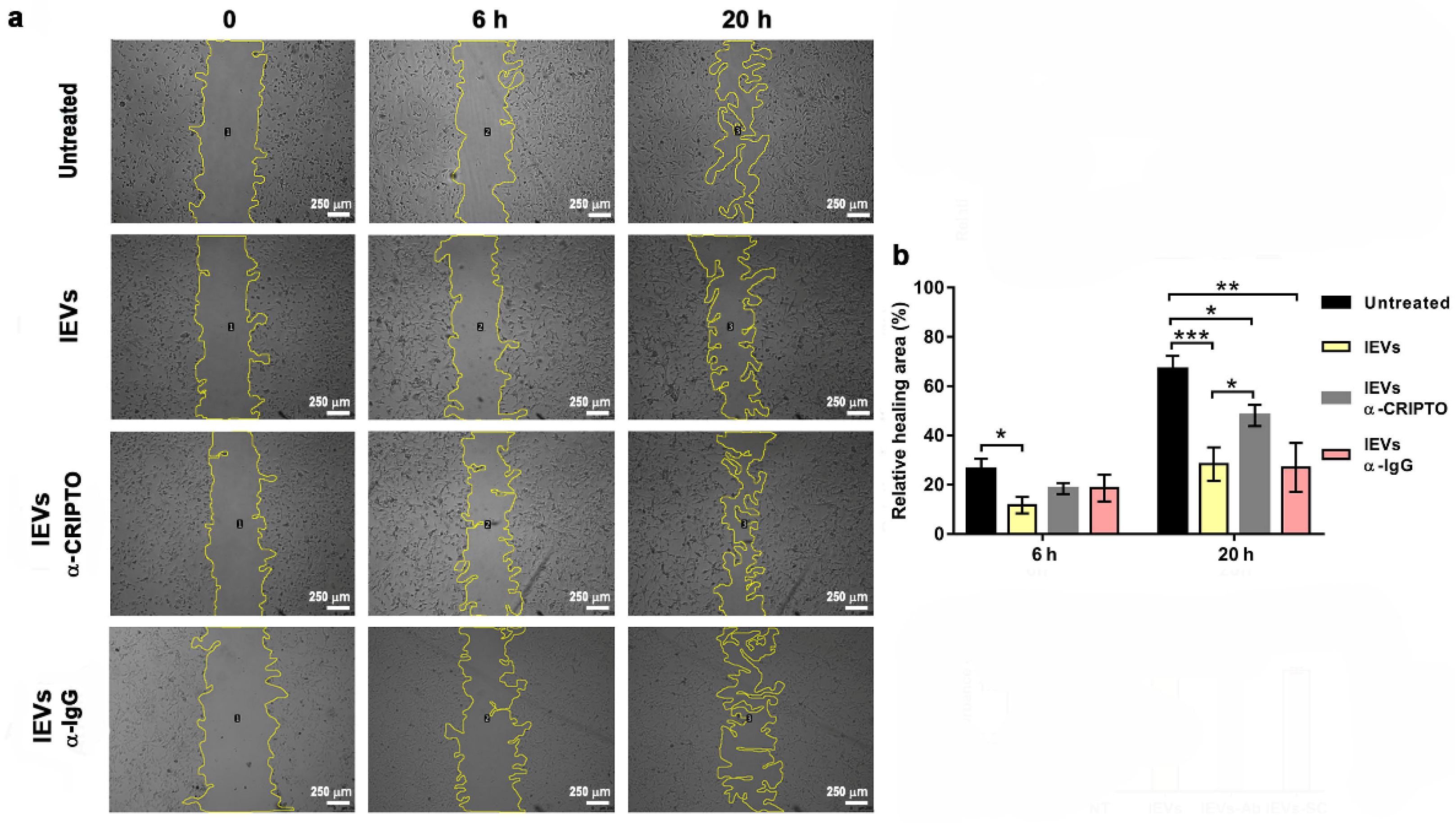
2.9. Wound Healing Assay
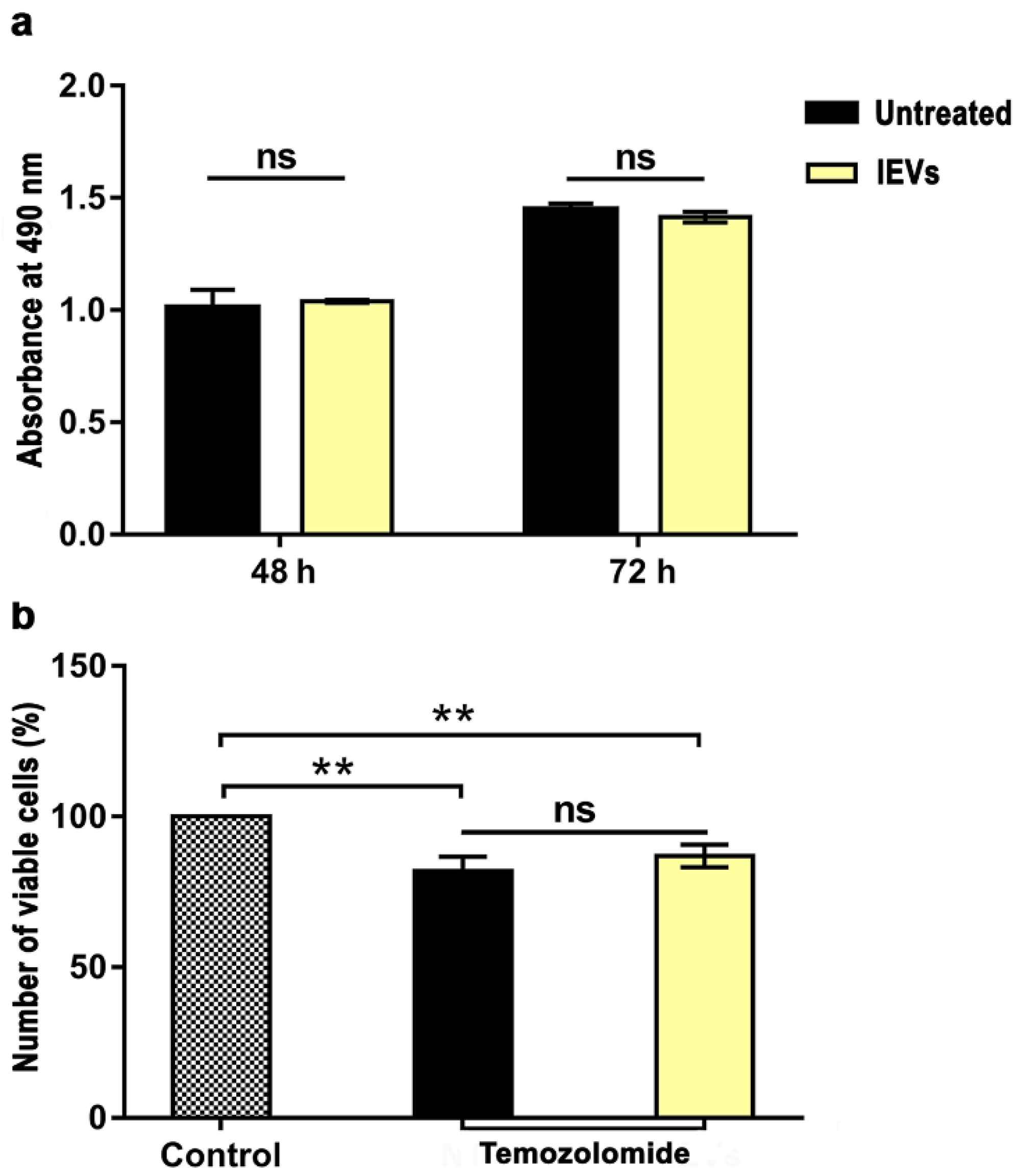
2.10. Cell Proliferation and Drug Sensitivity Assay
2.11. Quality Management
2.12. Statistical Analysis
3. Results
3.1. Isolation of Extracellular Vesicles from NTERA2 Teratocarcinoma Cells
3.2. Functional Characterization of NTERA2 Large Extracellular Vesicles
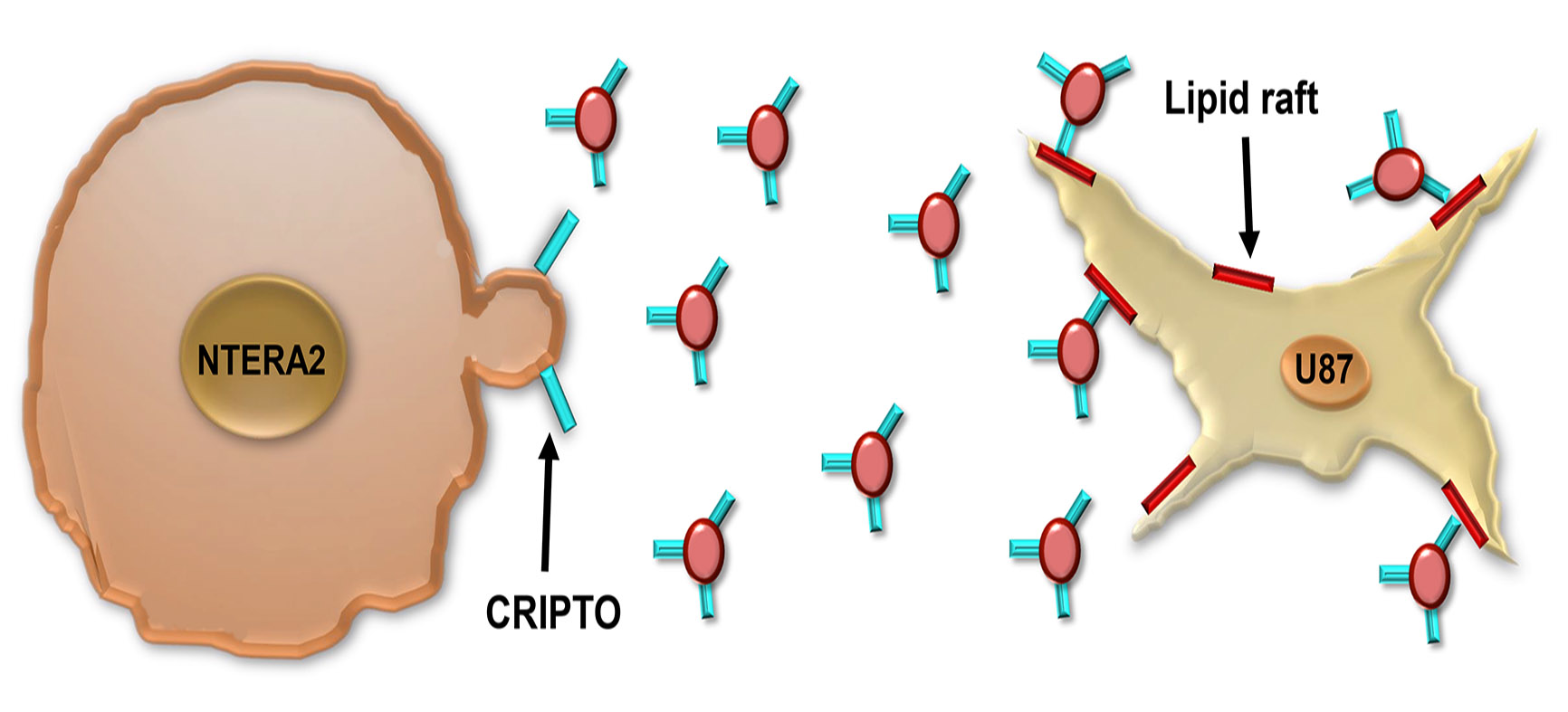
3.3. Association and Functional Relevance of CRIPTO in NTERA2 Extracellular Vesicles
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Mantile, F.; Franco, P.; Stoppelli, M.P.; Liguori, G.L. Biological role and clinical relevance of extracellular vesicles as key mediators of cell communication in cancer. In Biological Membrane Vesicles: Scientific, Biotechnological and Clinical Considerations. Advances in Biomembranes and Lipid Self-Assembly; Elsiever: Amsterdam, The Netherlands, 2020; Volume 33. [Google Scholar]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persico, M.G.; Liguori, G.L.; Parisi, S.; D’Andrea, D.; Salomon, D.S.; Minchiotti, G. Cripto in Tumors and Embryo Development. Biochim. Biophys. Acta Rev. Cancer 2001, 1552, S0304–S0419. [Google Scholar] [CrossRef]
- Minchiotti, G.; Parisi, S.; Liguori, G.L.; D’Andrea, D.; Persico, M.G. Role of the EGF-CFC Gene Cripto in Cell Differentiation and Embryo Development. Gene 2002, 287, S0378–S1119. [Google Scholar] [CrossRef]
- Ding, J.; Yang, L.; Yan, Y.T.; Chen, A.; Desai, N.; Wynshaw-Boris, A.; Shen, M.M. Cripto Is Required for Correct Orientation of the Anterior-Posterior Axis in the Mouse Embryo. Nature 1998, 395, 702–707. [Google Scholar] [CrossRef]
- Xu, C.; Liguori, G.; Persico, M.G.; Adamson, E.D. Abrogation of the Cripto Gene in Mouse Leads to Failure of Postgastrulation Morphogenesis and Lack of Differentiation of Cardiomyocytes. Development 1999, 126, 483–494. [Google Scholar] [CrossRef]
- Liguori, G.L.; Echevarría, D.; Improta, R.; Signore, M.; Adamson, E.; Martínez, S.; Persico, M.G. Anterior Neural Plate Regionalization in Cripto Null Mutant Mouse Embryos in the Absence of Node and Primitive Streak. Dev. Biol. 2003, 264, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Liguori, G.L.; Echevarria, D.; Bonilla, S.; D’andrea, D.; Liguoro, A.; Persico, M.G.; Martinez, S. Characterization of the functional properties of the neuroectoderm in mouse Cripto-/-embryos showing severe gastrulation defects. Int. J. Dev. Biol. 2004, 53, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Liguori, G.; Adamson, E.D.; Persico, M.G. Specific Arrest of Cardiogenesis in Cultured Embryonic Stem Cells Lacking Cripto-1. Dev. Biol. 1998, 196, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Parisi, S.; D’Andrea, D.; Lago, C.T.; Adamson, E.D.; Persico, M.G.; Minchiotti, G. Nodal-Dependent Cripto Signaling Promotes Cardiomyogenesis and Redirects the Neural Fate of Embryonic Stem Cells. J. Cell Biol. 2003, 163, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Gershon, E.; Hadas, R.; Elbaz, M.; Booker, E.; Muchnik, M.; Kleinjan-Elazary, A.; Karasenti, S.; Genin, O.; Cinnamon, Y.; Gray, P.C. Identification of Trophectoderm-Derived Cripto as an Essential Mediator of Embryo Implantation. Endocrinology 2018, 159, 1793–1807. [Google Scholar] [CrossRef]
- Freeman, D.W.; Sousa, E.R.; Karkampouna, S.; Zoni, E.; Gray, P.C.; Salomon, D.S.; Kruithof-de Julio, M.; Spike, B.T. Whence Cripto: The Reemergence of an Oncofetal Factor in ‘Wounds’ That Fail to Heal. Int. J. Mol. Sci. 2021, 22, 10164. [Google Scholar] [CrossRef]
- Ruggiero, D.; Nappo, S.; Nutile, T.; Sorice, R.; Talotta, F.; Giorgio, E.; Bellenguez, C.; Leutenegger, A.L.; Liguori, G.L.; Ciullo, M. Genetic Variants Modulating CRIPTO Serum Levels Identified by Genome-Wide Association Study in Cilento Isolates. PLoS Genet. 2015, 11, e1004976. [Google Scholar] [CrossRef] [Green Version]
- de Castro, N.P.; Rangel, M.C.; Nagaoka, T.; Salomon, D.S.; Bianco, C. Cripto-1: An Embryonic Gene That Promoted Tumorigeneis. Future Oncol. 2010, 6, 1127–1142. [Google Scholar] [CrossRef]
- Rodrigues Sousa, E.; Zoni, E.; Karkampouna, S.; La Manna, F.; Gray, P.C.; De Menna, M.; Kruithof-de Julio, M. A Multidisciplinary Review of the Roles of Cripto in the Scientific Literature through a Bibliometric Analysis of Its Biological Roles. Cancers 2020, 12, 1480. [Google Scholar] [CrossRef]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The Multifaceted Role of the Embryonic Gene Cripto-1 in Cancer, Stem Cells and Epithelial-Mesenchymal Transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Minchiotti, G.; Parisi, S.; Liguori, G.; Signore, M.; Lania, G.; Adamson, E.D.; Lago, C.T.; Persico, M.G. Membrane-Anchorage of Cripto Protein by Glycosylphosphatidylinositol and Its Distribution during Early Mouse Development. Mech. Dev. 2000, 90, S0925–S4773. [Google Scholar] [CrossRef]
- Chu, J.; Ding, J.; Jeays-Ward, K.; Price, S.M.; Placzek, M.; Shen, M.M. Non-Cell-Autonomous Role for Cripto in Axial Midline Formation during Vertebrate Embryogenesis. Development 2005, 132, 5539–5551. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhang, Y.; Zhang, M.; Li, T.; Zheng, X.; Guo, Q.; Zhang, X. Exosomal Cripto-1 Serves as a Potential Biomarker for Perihilar Cholangiocarcinoma. Front. Oncol. 2021, 11, 3144. [Google Scholar] [CrossRef]
- Ciccodicola, A.; Dono, R.; Obici, S.; Simeone, A.; Zollo, M.; Persico, M.G. Molecular Characterization of a Gene of the ‘EGF Family’ Expressed in Undifferentiated Human NTERA2 Teratocarcinoma Cells. EMBO J. 1989, 8, 1987–1991. [Google Scholar] [CrossRef]
- Dono, R.; Montuori, N.; Rocchi, M.; De Ponti-Zilli, L.; Ciccodicola, A.; Persico, M.G. Isolation and Characterization of the CRIPTO Autosomal Gene and Its X-Linked Related Sequence. Am. J. Hum. Genet. 1991, 49, 555–565. [Google Scholar]
- Andrews, P.W. Teratocarcinomas and Human Embryology: Pluripotent Human EC Cell Lines. Review Article. Apmis 1998, 106, 158–168. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Molecular Sciences Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant Astrocytomas Originate from Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Altmann, C.; Keller, S.; Schmidt, M.H.H. The Role of SVZ Stem Cells in Glioblastoma. Cancers 2019, 11, 448. [Google Scholar] [CrossRef] [Green Version]
- Almengló, C.; Caamaño, P.; Fraga, M.; Devesa, J.; Costoya, J.A.; Arce, V.M.; José Costoya, C.A. From Neural Stem Cells to Glioblastoma: A Natural History of GBM Recapitulated in Vitro. J. Cell Physiol. 2021, 236, 7390–7404. [Google Scholar] [CrossRef]
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural Stem Cells Target Intracranial Glioma to Deliver an Oncolytic Adenovirus in Vivo. Gene Ther. 2009, 16, 262–278. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhong, Q.; Liu, H.; Liu, P.; Wu, J.; Ma, D.; Chen, X.; Yang, X. Conditioned Medium from Neural Stem Cells Inhibits Glioma Cell Growth. Cell Mol. Biol. 2016, 62, 68–73. [Google Scholar] [CrossRef]
- Attia, N.; Mashal, M.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Cationic Niosome-Based HBMP7 Gene Transfection of Neuronal Precursor NT2 Cells to Reduce the Migration of Glioma Cells in Vitro. J. Drug Deliv. Sci. Technol. 2019, 53, 101219. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S. Human NT2 Neural Precursor-Derived Tumor-Infiltrating Cells as Delivery Vehicles for Treatment of Glioblastoma. Hum. Gene Ther. 2010, 21, 683–694. [Google Scholar] [CrossRef]
- Romancino, D.P.; Paterniti, G.; Campos, Y.; De Luca, A.; Di Felice, V.; D’azzo, A.; Bongiovanni, A. Identification and Characterization of the Nano-Sized Vesicles Released by Muscle Cells. FEBS Lett. 2013, 587, 1379–1384. [Google Scholar] [CrossRef] [Green Version]
- Lešer, V.; Drobne, D.; Pipan, Ž.; Milani, M.; Tatti, F. Comparison of Different Preparation Methods of Biological Samples for FIB Milling and SEM Investigation. J. Microsc. 2009, 233, 309–319. [Google Scholar] [CrossRef]
- Liguori, G.L.; Kisslinger, A. Standardization and reproducibility in EV research: The support of a quality management system. In Biological Membrane Vesicles: Scientific, Biotechnological and Clinical Considerations. Advances in Biomembranes and Lipid Self-Assembly; Elsiever: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Bongiovanni, A.; Colotti, G.; Liguori, G.L.; Di Carlo, M.; Digilio, F.A.; Lacerra, G.; Mascia, A.; Cirafici, A.M.; Barra, A.; Lanati, A.; et al. Applying Quality and Project Management Methodologies in Biomedical Research Laboratories: A Public Research Network’s Case Study. Accredit. Qual. Assur. 2015, 20, 203–213. [Google Scholar] [CrossRef]
- Digilio, F.A.; Lanati, A.; Bongiovanni, A.; Mascia, A.; Di Carlo, M.; Barra, A.; Cirafici, A.M.; Colotti, G.; Kisslinger, A.; Lacerra, G.; et al. Quality-Based Model for Life Sciences Research Guidelines. Accredit. Qual. Assur. 2016, 21, 221–230. [Google Scholar] [CrossRef]
- Yekula, A.; Minciacchi, V.R.; Morello, M.; Shao, H.; Park, Y.; Zhang, X.; Muralidharan, K.; Freeman, M.R.; Weissleder, R.; Lee, H.; et al. Large and Small Extracellular Vesicles Released by Glioma Cells in Vitro and in Vivo. J. Extracell. Vesicles 2020, 9, 1689784. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.J.; Homer, N.; O’Connor, B.D.; Chen, Z.; Eskin, A.; Lee, H.; Merriman, B.; Nelson, S.F. U87MG Decoded: The Genomic Sequence of a Cytogenetically Aberrant Human Cancer Cell Line. PLoS Genet. 2010, 6, e1000832. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.P.; Van Meerlo, J.; Assaraf, Y.G.; et al. Exosomes Secreted by Apoptosis-Resistant Acute Myeloid Leukemia (AML) Blasts Harbor Regulatory Network Proteins Potentially Involved in Antagonism of Apoptosis. Mol. Cell. Proteom. 2016, 15, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; De Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In Vivo Imaging Reveals Extracellular Vesicle-Mediated Phenocopying of Metastatic Behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D.; Sciences, B.; Angeles, L.; Diseases, U.; Children, B. The Emerging Role of Large Oncosomes. Semin. Cell Dev. Biol. 2016, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active Endocannabinoids Are Secreted on Extracellular Membrane Vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Denzer, K.; van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular Dendritic Cells Carry MHC Class II-Expressing Microvesicles at Their Surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Prada, I.; Amin, L.; Furlan, R.; Legname, G.; Verderio, C.; Cojoc, D. A New Approach to Follow a Single Extracellular Vesicle–Cell Interaction Using Optical Tweezers. Biotechniques 2016, 60, 35. [Google Scholar] [CrossRef] [Green Version]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes Surf on Filopodia to Enter Cells at Endocytic Hot Spots, Traffic within Endosomes, and Are Targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular Vesicles from Blood Plasma: Determination of Their Morphology, Size, Phenotype and Concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Höög, J.L.; Lötvall, J. Diversity of extracellular vesicles in human ejaculates revealed by cryo-electron microscopy. J. Extracell. Vesicles 2015, 4, 28680. [Google Scholar] [CrossRef] [Green Version]
- Zabeo, D.; Cvjetkovic, A.; Lässer, C.; Schorb, M.; Lötvall, J.; Höög, J.L. Exosomes purified from a single cell type have diverse morphology. J. Extracell. Vesicles 2017, 6, 1329476. [Google Scholar] [CrossRef] [Green Version]
- Dubochet, J.; Adrian, M.; Chang, J.-J.; Homo, J.-C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef] [Green Version]
- Kralj-Iglič, V.; Pocsfalvi, G.; Mesarec, L.; Šuštar, V.; Hägerstrand, H.; Iglič, A. Minimizing isotropic and deviatoric membrane energy—An unifying formation mechanism of different cellular membrane nanovesicle types. PLoS ONE 2020, 15, e0244796. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Bianco, C.; Strizzi, L.; Hamada, S.; Mancino, M.; Bailly, V.; Mo, W.; Wen, D.; Miatkowski, K.; Gonzales, M.; et al. Growth Factor Induction of Cripto-1 Shedding by Glycosylphosphatidylinositol-Phospholipase D and Enhancement of Endothelial Cell Migration. J. Biol. Chem. 2007, 282, 31643–31655. [Google Scholar] [CrossRef] [Green Version]
- Hägerstrand, H.; Mrówczyńska, L.; Salzer, U.; Prohaska, R.; Michelsen, K.A.; Kralj-Iglič, V.; Iglič, A. Curvature-dependent lateral distribution of raft markers in the human erythrocyte membrane. Mol. Membr. Biol. 2006, 23, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Pollet, H.; Conrard, L.; Cloos, A.S.; Tyteca, D. Plasma membrane lipid domains as platforms for vesicle biogenesis and shedding? Biomolecules 2018, 8, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, M. Exosomes and GPI-Anchored Proteins: Judicious Pairs for Investigating Biomarkers from Body Fluids. Adv. Drug Deliv. Rev. 2020, 161, 110–123. [Google Scholar] [CrossRef]
- Wechselberger, C.; Ebert, A.D.; Bianco, C.; Khan, N.I.; Sun, Y.; Wallace-Jones, B.; Montesano, R.; Salomon, D.S. Cripto-1 Enhances Migration and Branching Morphogenesis of Mouse Mammary Epithelial Cells. Exp. Cell Res. 2001, 266, 95–105. [Google Scholar] [CrossRef]
- Bianco, C.; Adkins, H.B.; Wechselberger, C.; Seno, M.; Normanno, N.; De Luca, A.; Sun, Y.; Khan, N.; Kenney, N.; Ebert, A.; et al. Cripto-1 Activates Nodal- and ALK4-Dependent and -Independent Signaling Pathways in Mammary Epithelial Cells. Mol. Cell. Biol. 2002, 22, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Hamada, S.; Bianco, C.; Mancino, M.; Nagaoka, T.; Gonzales, M.; Bailly, V.; Strizzi, L.; Salomon, D.S. Requirement of Glycosylphosphatidylinositol Anchor of Cripto-1 for Trans Activity as a Nodal Co-Receptor. J. Biol. Chem. 2007, 282, 35772–35786. [Google Scholar] [CrossRef] [Green Version]
- Bianco, C.; Rangel, M.C.; Castro, N.P.; Nagaoka, T.; Rollman, K.; Gonzales, M.; Salomon, D.S. Role of Cripto-1 in Stem Cell Maintenance and Malignant Progression. Am. J. Pathol. 2010, 177, 532–540. [Google Scholar] [CrossRef]
- Giorgio, E.; Liguoro, A.; D’Orsi, L.; Mancinelli, S.; Barbieri, A.; Palma, G.; Arra, C.; Liguori, G.L. Cripto haploinsufficiency affects in vivo colon tumor development. Int. J. Oncol. 2014, 45, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Pilgaard, L.; Mortensen, J.H.; Henriksen, M.; Olesen, P.; Sørensen, P.; Laursen, R.; Vyberg, M.; Agger, R.; Zachar, V.; Moos, T.; et al. Cripto-1 Expression in Glioblastoma Multiforme. Brain Pathol. 2014, 24, 360–370. [Google Scholar] [CrossRef]
- Tysnes, B.B.; Sætran, H.A.; Mørk, S.J.; Margaryan, N.V.; Eide, G.E.; Petersen, K.; Strizzi, L.; Hendrix, M.J. Age-Dependent Association between Protein Expression of the Embryonic Stem Cell Marker Cripto-1 and Survival of Glioblastoma Patients. Transl. Oncol. 2013, 6, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Alowaidi, F.; Hashimi, S.M.; Nguyen, M.; Meshram, M.; Alqurashi, N.; Cavanagh, B.L.; Bellette, B.; Ivanovski, S.; Meedenyia, A.; Wood, S.A. Investigating the Role of CRIPTO-1 (TDGF-1) in Glioblastoma Multiforme U87 Cell Line. J. Cell. Biochem. 2019, 120, 7412–7427. [Google Scholar] [CrossRef]
- Alowaidi, F.; Hashimi, S.M.; AlQurashi, N.; Wood, S.A.; Wei, M.Q. Cripto-1 overexpression in U87 glioblastoma cells activates MAPK, focal adhesion and ErbB pathways. Oncol. Lett. 2019, 18, 3399–3406. [Google Scholar] [CrossRef] [Green Version]
- Gudbergsson, J.M.; Duroux, M. Cripto-1 Localizes to Dynamic and Shed Filopodia Associated with Cellular Migration in Glioblastoma Cells. Eur. J. Cell Biol. 2019, 98, 151044. [Google Scholar] [CrossRef]
- Kralj-Iglic, V. Stability of Membranous Nanostructures: A Possible Key Mechanism in Cancer Progression. Int. J. Nanomed. 2012, 7, 3579–3596. [Google Scholar] [CrossRef] [Green Version]
- De Silva, T.; Ye, G.; Liang, Y.Y.; Fu, G.; Xu, G.; Peng, C. Nodal Promotes Glioblastoma Cell Growth. Front. Endocrinol. 2012, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Su, G.; Meyer, K.; Nandini, C.D.; Qiao, D.; Salamat, S.; Friedl, A. Glypican-1 Is Frequently Overexpressed in Human Gliomas and Enhances FGF-2 Signaling in Glioma Cells. Am. J. Pathol. 2006, 168, 2014–2026. [Google Scholar] [CrossRef] [Green Version]
- Bu, G.; Maksymovitch, E.A.; Geuze, H.; Schwartz, A.L. Subcellular Localization and Endocytic Function of Low Density Lipoprotein Receptor-Related Protein in Human Glioblastoma Cells. J. Biol. Chem. 1994, 269, 29874–29882. [Google Scholar] [CrossRef]
- Kim, H.S.; Park, Y.H.; Lee, H.S.; Kwon, M.J.; Song, J.H.; Chang, I.B. Propranolol Inhibits the Proliferation of Human Glioblas-Toma Cell Lines through Notch1 and Hes1 Signaling System. J. Korean Neurosurg. Soc. 2021, 64, 716–725. [Google Scholar] [CrossRef]
- Liu, K.; Tsung, K.; Attenello, F.J. Characterizing Cell Stress and GRP78 in Glioma to Enhance Tumor Treatment. Front. Oncol. 2020, 10, 608911. [Google Scholar] [CrossRef]
- Pardali, K.; Moustakas, A. Actions of TGF-β as Tumor Suppressor and pro-Metastatic Factor in Human Cancer. Biochim. Biophys. Acta Rev. Cancer 2007, 1775, 21–62. [Google Scholar] [CrossRef]
- Sun, G.; Shi, L.; Li, M.; Jiang, N.; Fu, L.; Guo, J. Lefty Inhibits Glioma Growth by Suppressing Nodal-Activated Smad and ERK1/2 Pathways. J. Neurol. Sci. 2014, 347, 137–142. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Chino, H.; Akiya, M.; Hashimura, M.; Yokoi, A.; Tochimoto, M.; Nakagawa, M.; Jiang, Z.; Saegusa, M. Requirements of LEFTY and Nodal Overexpression for Tumor Cell Survival under Hypoxia in Glioblastoma. Mol. Carcinog. 2020, 59, 1409–1419. [Google Scholar] [CrossRef]
- Rodrigues-Junior, D.M.; Tsirigoti, C.; Lim, S.K.; Heldin, C.-H.; Moustakas, A. Extracellular Vesicles and Transforming Growth Factor β Signaling in Cancer. Front. Cell Dev. Biol. 2022, 10, 761. [Google Scholar] [CrossRef]
- Schneider, M.; Winkler, K.; Kell, R.; Pfaffl, M.W.; Atkinson, M.J.; Moertl, S. The Chaperone Protein GRP78 Promotes Survival and Migration of Head and Neck Cancer After Direct Radiation Exposure and Extracellular Vesicle-Transfer. Front. Oncol. 2022, 12, 842418. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantile, F.; Kisovec, M.; Adamo, G.; Romancino, D.P.; Hočevar, M.; Božič, D.; Bedina Zavec, A.; Podobnik, M.; Stoppelli, M.P.; Kisslinger, A.; et al. A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications. Cancers 2022, 14, 3700. https://doi.org/10.3390/cancers14153700
Mantile F, Kisovec M, Adamo G, Romancino DP, Hočevar M, Božič D, Bedina Zavec A, Podobnik M, Stoppelli MP, Kisslinger A, et al. A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications. Cancers. 2022; 14(15):3700. https://doi.org/10.3390/cancers14153700
Chicago/Turabian StyleMantile, Francesca, Matic Kisovec, Giorgia Adamo, Daniele P. Romancino, Matej Hočevar, Darja Božič, Apolonija Bedina Zavec, Marjetka Podobnik, Maria Patrizia Stoppelli, Annamaria Kisslinger, and et al. 2022. "A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications" Cancers 14, no. 15: 3700. https://doi.org/10.3390/cancers14153700