1. Introduction
Lung tumors are among the most common in the world and are one of the leading causes of cancer deaths worldwide [
1]. Among them, NSCLC accounts for 85% of all lung tumor cases and it is mainly composed of lung squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD) [
1]. Although the available therapeutic arsenal, including surgical procedures, chemotherapy, targeted molecules and immunotherapy, have allowed undeniable progress in lung cancer treatment, the 5-year survival rate of NSCLC patients remains unsatisfactory. This relative inefficiency is mainly due to the lack of information about the tumor microenvironment during treatment decision. For this reason, many investigations have focused on the discovery of a new prognostic assessment method to help individualized treatment of NSLCL patients [
2].
Autophagy is a cellular process associated with the prevalence and progression of lung cancer [
3,
4]. Autophagy is a conserved catabolism pathway that plays a key role in the maintenance of cellular homeostasis. It is a multistep mechanism, consisting of the formation of the phagophore that elongates and engulfs targeted proteins or organelles in a double-membrane vesicle called the autophagosome, and finally fuses with late endosomes and/or lysosomes [
5]. This process is orchestrated by a large variety of proteins, including the autophagic proteins (Atg), organized in complexes. Autophagy induction is modulated by two protein complexes, the ULK1/2 (unc51-like autophagy activating kinase) and the Beclin-1/PI3KC3 (class III phosphatidylinositol 3-kinase) complexes. Once activated, these complexes recruit other proteins involved in the elongation and formation of autophagosomes, including the two conjugated systems Atg12-Atg5-Atg16L and LC3. After completion, the mature autophagosome fuses with lysosomes to form autolysosomes, wherein the sequestered materials and organelles are degraded by lysosomal enzymes [
6]. Then, the degradation products are recycled for cell synthesis biological processes. Autophagy is one of the most important survival mechanisms under stress conditions and is involved in cellular homeostasis and proliferation [
5]. Several studies have demonstrated links between autophagy and carcinogenesis, highlighting a dual role for autophagy in cancer. Depending on the tumor model and/or tumor state, autophagy may have pro- or anti-tumor effects. In the initial stage of cancer, autophagy protects normal cells from tumorigenesis by preventing DNA damages and mutations [
3]. In established solid tumors, autophagy has been shown to favor tumor development by enhancing tumor growth, cell survival, resistance to platinum-based chemotherapy and metastasis formation [
7]. Autophagy may also interfere with immunotherapy, since some studies showed a link between autophagy and immune checkpoint activity and/or expression, including CTLA-4, IDO and PD1/PD-L1 [
8,
9]. Autophagy also has a critical function in tumor immune cells and tumor immune response, promoting the immunogenic cell death of tumor cells and favoring immune cell activation and proliferation [
10]. Meanwhile, autophagy in cancer-associated fibroblasts (CAFs) promotes tumorigenesis by providing nutrients to the cancerous cells and by favoring epithelial to mesenchymal transition, angiogenesis and stemness [
11].
Many transcriptomic analyses performed on autophagy genes have focused on the discovery of new biomarkers to predict the efficiency of anti-tumor therapies and to guide individualized treatment in NSCLC patients [
12,
13,
14,
15,
16]. However, the majority of these studies are based on global transcriptomic analysis of the whole tumor microenvironment, and few investigations have been carried out on malignant cells themselves. Regarding the global effect of autophagy on cells infiltrating the tumor microenvironment, it is important to determine a signature to identify the functional status of each cell type. In this study, we explore the relationship between 232 autophagy-related genes and biological pathways related to tumor progression in multiple LUAD datasets. Comparing tumors with adjacent tissue, we identified two signatures composed of twenty-three (signature A) and twelve (signature B) genes, and these signatures were correlated with survival, tumor metabolic status and immunology factors in LUAD patients. RNA sequence profiling of flow-sorted malignant cells, endothelial cells, immune cells and fibroblasts from freshly resected primary human NSCLC reveals that signature B was mainly expressed by malignant cells. The predominant expression of signature B in malignant cells was validated in the single cell sequencing data analysis. Deeper investigations supported the correlation between autophagy with tumor cell proliferation and immune checkpoint expression in malignant cells, highlighting the impact of autophagy in tumor cell progression and its potential role in immunotherapy. Therefore, our study provides a new autophagy-related signature that predicts the biological status of malignant cells in LUAD patients.
2. Materials and Methods
2.1. Dataset Source, Pre-Processing and Workflow
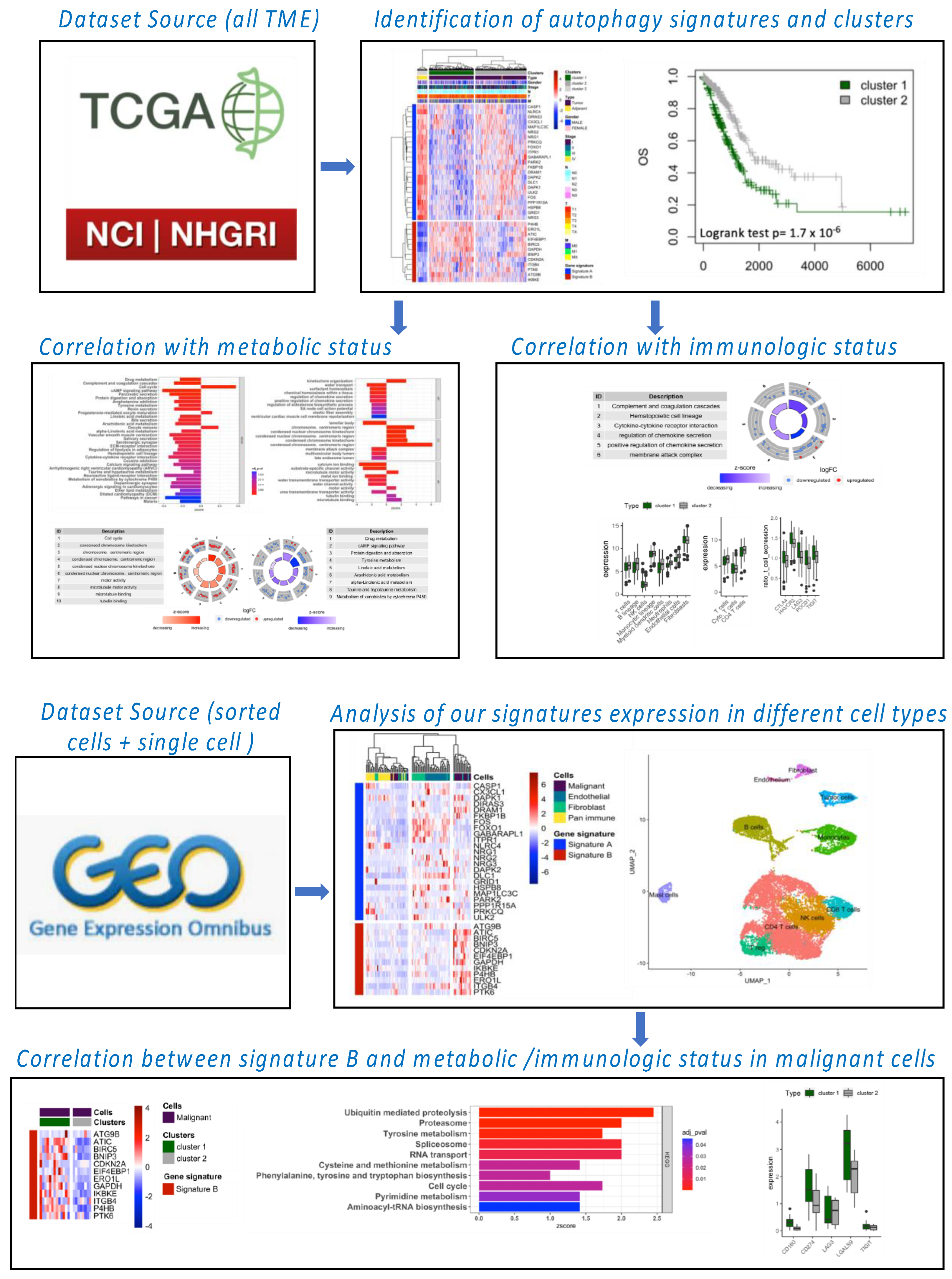
The workflow of our bioinformatic analysis is summarized in
Figure 1. LUNG and LUAD gene expression datasets and associated clinical information were obtained from The Cancer Genome Atlas (TCGA) and Gene-Expression Omnibus (GEO) databases. The download gene expression profiles from TCGA met the following conditions: (1) the primary site was “bronchus and lung”; (2) the program was “TCGA”; (3) the disease type was “adenomas and adenocarcinomas” and/or “squamous cell neoplasms”; (4) the data category was “transcriptome profiling”; (5) the data type was “Gene Expression Quantification”; and (6) the workflow type was “HTSeq-FPKM”. TCGA-LUNG samples used for this analysis included 110 normal samples and 1019 tumor samples and TCGA-LUAD included 59 normal samples and 517 tumor samples. In addition, we downloaded series matrix files and platform files of four datasets, including one for global TME (GSE31210), one for RNA sequence profiling of flow-sorted malignant cells, endothelial cells, immune cells and fibroblasts from resected primary human NSCLC (GSE111907), one for single-cell analysis (GSE123904) and one for A549 cells invalidated by siRNA for atg5 and ulk1 genes involved in the autophagy process (GSE73158). The basic information regarding all databases is provided in
Table 1.
The fragments per kilobase million (FPKM) values were converted into the transcripts per million (TPM) data using the R package “limma”. R (version 4.0.3, R core team,
https://www.r-project.org, accessed on 5 June 2022) was used to process data. Processing for GSE datasets will be explained in the appropriate section.
2.2. Autophagy Signature and Clustering Analysis
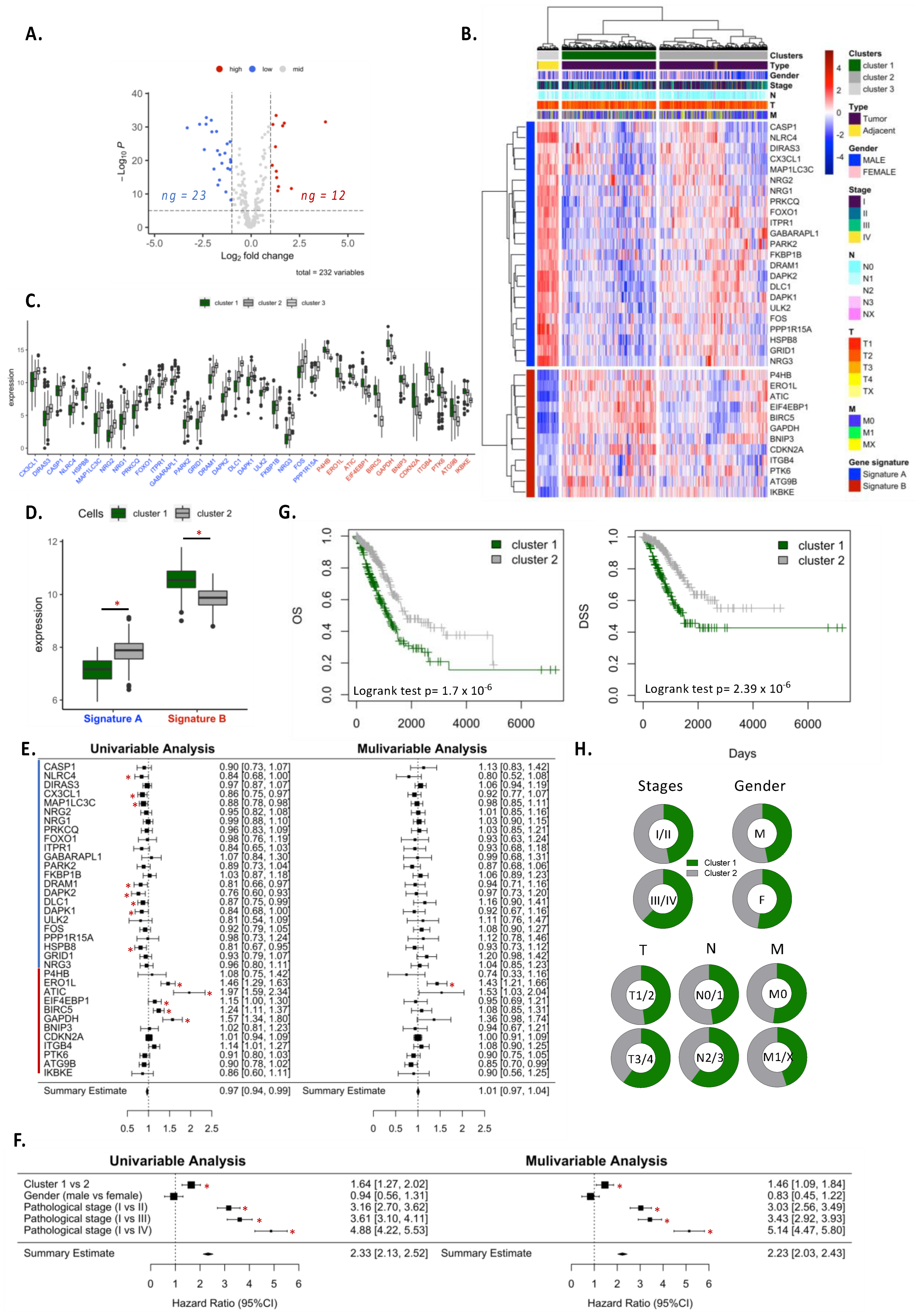
We used the human autophagy database (HADd) to analyze the differential expression of autophagy-related genes (n = 232) between normal and tumor tissue. Differentially expressed genes were based on logFC > 1 or <−1 and adjusted p-value < 0.05 using “limma” package in R. LUNG or LUAD samples were grouped into clusters according to their expression of autophagy signature genes (genes differentially expressed between adjacent and tumor samples). Kaplan–Meier survival curves were applied to each cluster and log-rank tests were performed to compare the overall survival (OS) and disease-specific survival (DSS) between clusters. Univariate and multivariate Cox analysis was performed to analyze the hazard ratio of clusters. Similar analysis was performed for individual genes of autophagy signature.
2.3. Functional Annotation Enrichment
To determine the variation of biological pathways between clusters, the differential expression analysis of whole genome between two clusters was performed. Differentially expressed genes were based on logFC > 1 or <−1 and adjusted
p-value < 0.05 using “limma” package in R. Based on DEG analyses, gene set enrichment analysis (GSEA) and Gene Ontology (GO) enrichment analysis were performed using the “Enrichr” website (
https://maayanlab.cloud/Enrichr/, accessed on 5 June 2022) and the results were plotted using the “GOplot” R package. We selected the function and pathways with a strict
p-value < 0.05. For “circle plot”, we selected important pathways or functions involved in anabolism or catabolism.
2.4. Immune Cell Infiltration, Stromal Cell Population and Exhaustion Marker Expression Analysis
For immune cell infiltration and the stromal cell population, we applied the microenvironment cell population-counter (MCP-count) method [
17]. A total of 10 cell signatures were calculated to determine T cells, cytotoxic T cells, CD4+ T cells, B cell lineage, NK cells, monocyte lineage, myeloid dendritic cells, neutrophils, endothelial cells and fibroblasts. To determine the expression of exhaustion markers in T cells, we calculated the expression of
CTLA-4,
HAVCR2,
LAG3,
PDCD1 and
TIGIT genes according to the median of T-cell expression in TCGA.
2.5. Autophagy Signature Expression in Sorted-Cell Fresh Tumor Samples Datasets and Single Cell Analysis
RNA sequence profiling of flow-sorted malignant cells (EPCAM+ CD45− CD31−), endothelial cells (CD31+ CD45− EPCAM−), immune cells (CD45+ EPCAM−) and fibroblasts (CD10+ CD45− EPCAM− CD31− CD10+) from freshly resected primary human NSCLC (GSE111907 dataset) were used to calculated expression levels of autophagy signature A and B on different cell populations. For these analyses, we only selected adenocarcinoma subtypes. We analyzed 21 malignant samples, 22 pan-immune samples, 23 endothelial samples and 22 fibroblasts samples. Samples were clustered according to their expression of autophagy signature gene and data were visualized by “pheatmap” R package. For single-cell analysis, we utilized GSE123904 datasets. We used only primary tumor samples for this analysis (LX653, LX661, LX675, LX676, LX678, LX680, LX682 and LX684). A total of 18,124 cells were analyzed. Single cell clustering and dimension reduction were performed by R package “Seurat”. The principal component analysis (PCA), “FindNeighbors” and “FindClusters” packages were employed to construct the cell culturing. The “UMAP” package was used to visualize data, and we utilized the “FeaturePlot” function form “Seurat” to visualize the expression of the autophagy signatures. Cell clusters were annotated according to the gene expression in each cluster revealed by the “FindAllMarkers” function of “Seurat” package.
2.6. Autophagy Clustering Analysis in Malignant Tumor Cells
We selected 22 malignant samples (adenocarcinoma) from the GSE111907 dataset and samples were clustered according to their expression of the autophagy signature B. Gene Set Enrichment Analysis (GSEA) was performed as previously described.
2.7. Gene Set Enrichment Analysis in A549 Deficient for Autophagy
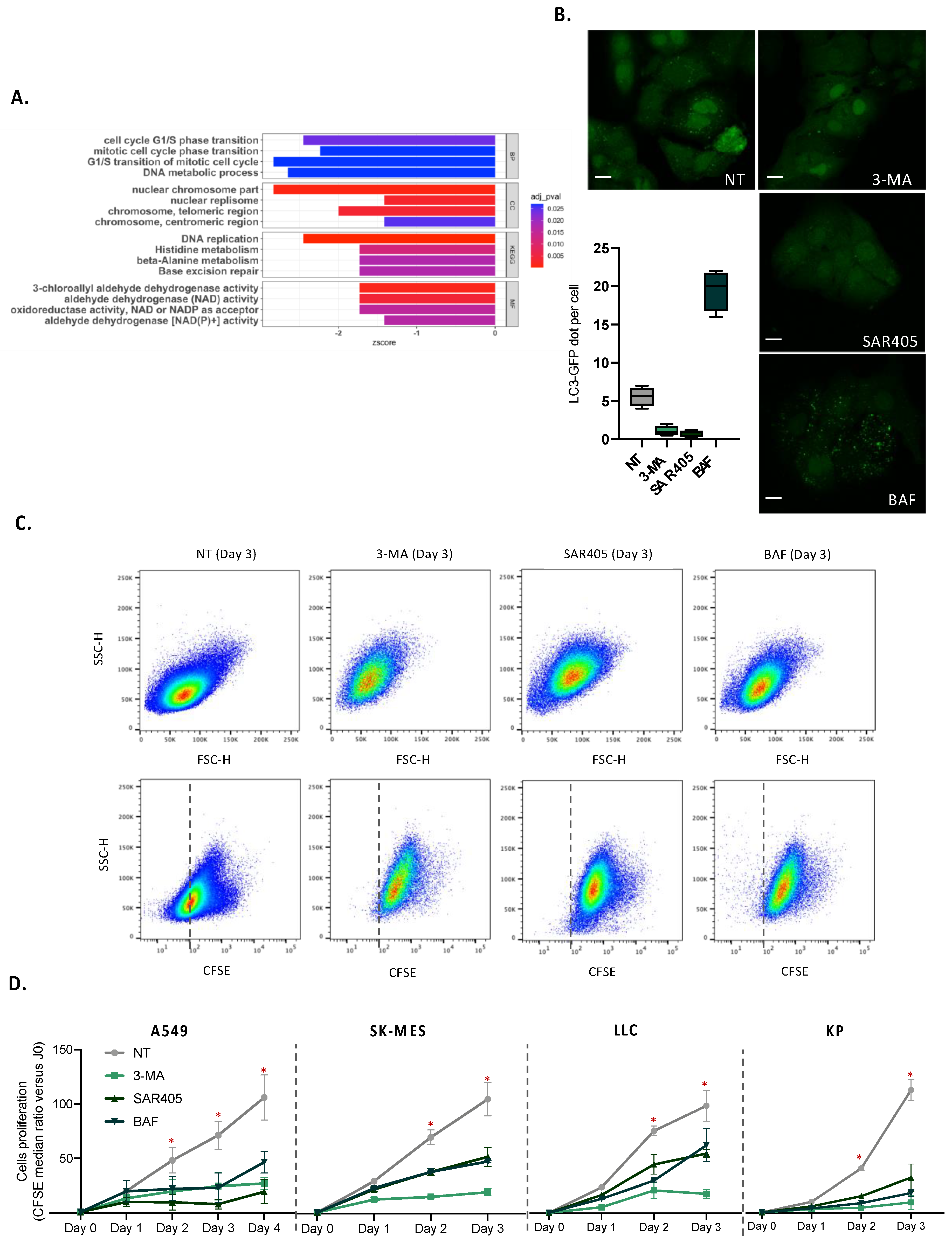
To examine the impact of autophagy genes in global biological processes, we used GSE73158 datasets. We selected three lung adenocarcinoma cell lines, A549 treated with siRNA against ATG5, A549 treated with siRNA against ULK1, and their respective siRNA control. We performed differential expression analysis for siRNA-treated cells according to their respective control and selected upregulated (logFC > 1) or down-regulated (logFC < −1) genes. To perform functional annotation enrichment analyses, we chose genes which were significantly modified in both siATG5- and siULK1-treated cells.
2.8. Cell Culture, Proliferation Analysis and Confocal Microscopy
The human lung adenocarcinoma A549 cell line and the murine adenocarcinoma LLC and carcinoma KP cell lines were cultured in DMEM F-12 medium (Gibco, Waltham, MA, USA) supplemented with 10% FBS (Eurobio Scientific, Ulis, France), 1% non-essential amino acid (Gibco), 1% herpes (Gibco), 1% glutamate (Gibco) and 1% Na+/pyruvate (Gibco) in a standard 5% CO2 incubation atmosphere at 37 °C. The human lung squamous cell carcinoma SK-MES cell line was cultured in EMEM F-12 medium (Gibco) supplemented with 10% FBS (Eurobio Scientific), 1% non-essential amino acid (Gibco), 1% 1% herpes (Gibco), 1% glutamate (Gibco) and 1% Na+/pyruvate (Gibco) in a standard 5% CO2 incubation atmosphere at 37 °C.
For in vitro proliferation assays, 150,000 cells were stained with CFSE (1/500, ThermoFischer, Waltham, MA, USA) for 30 min at 37 °C in PBS and plated in the 6-well plate for 24 h. Cells were cultured for 24, 48, 72 and 96 h in the presence or not of 10 mM of 3-methyladenin (Sigma, Saint-Louis, MO, USA), 100 nM of wortmannin (Sigma), 10 µM of SAR405 (MedChemExpress, Monmouth Junction, NJ, USA), or 100 nM of bafilomycin (Sigma), and stained with live/dead kit (1/100, near-IR, ThermoFisher). Analysis of CSFE staining was performed using the BD LSR Fortessa Cell analyzer. Flow cytometry data were analyzed by FlowJo software.
For in vitro analysis of the autophagy level, 35,000 A549 cells expressing the GFP-LC3 protein were plated in a 24-well plate containing coverslips for 24 h. Cells were then cultured for 24 h in the presence or not of 10 mM of 3-methyladenin (Sigma), 100 nM of wortmannin (Sigma), 10 µM of SAR405 (MedChemExpress), or 100 nM of bafilomycin (Sigma), and were mounted on the slides using glycergel (Dako, Santa Clara, CA, USA). The autophagosomes were observed by confocal microscopy (LSM 710) and enumerated by a personal R script.
2.9. Statistical Analysis
R software (v4.0.3) was used for all bioinformatic statistical analyses, and PRISM software was employed for in vitro experiments. The Wilcoxon test was used to compare the differences between the two groups. The Kruskal–Wallis test was utilized to compare the differences between three groups and above. The survival time of the patient was evaluated by Kaplan–Meier survival analysis, and the different groups were compared by utilizing a log-rank test. Univariate and multivariate Cox regression analysis was used to investigate the independent prognostic factor, employing the “survival” R package. The Benjamin–Hochberg method was used to calculate p_value for FRDs conversation and DEG analyses. Single-cell analysis was performed using R package “Seurat”. Survival curves were performed utilizing R package “survminer”. All heatmaps were generated by R package “pheatmap”. We employed “GOplot” R package to visualize the functional annotation enrichment analyses. Data visualization was performed using R package “ggplot2”. The R packages utilized in this study could be obtained from “bioconduction”.
4. Discussion
As a central process of self-digestion and stress adaptation, autophagy has a remarkable impact on tumor development [
4,
7,
18]. It can provide nutrients for cancer cell survival, proliferation and migration, promotes drug resistance and helps tumor cells to evade immune surveillance [
4]. In lung cancers, several studies showed that autophagy promotes tumor cell growth and resistance to radiation or chemotherapy [
19,
20]. However, due to the difficulty of visualizing and quantifying autophagy in tumor patients, the role of autophagy in NSCLC patients is still unclear [
20,
21].
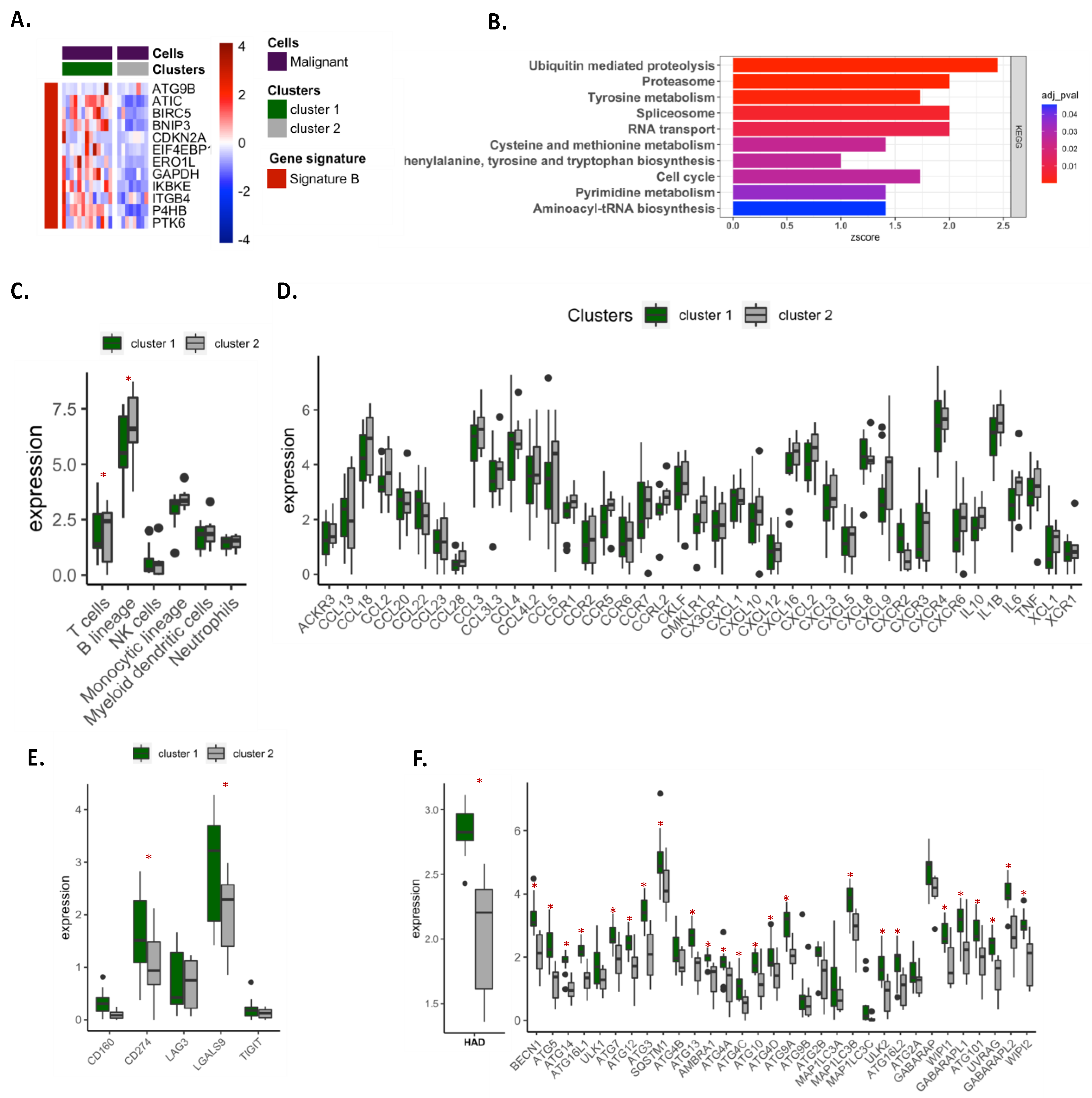
Gene expression analysis appears to be a relevant approach to analyze autophagy in NSCLC patients. We first conducted our analysis in LUAD and LUSC, which accounted for the majority of NSCLC. Based on the TCGA database, our preliminary exploration revealed that modification for the expression of autophagy genes can be observed in tumor samples, demonstrating that autophagy is particularly active in cancers. Autophagy expression was very dependent on cancer subtypes, and clear clustering of patients has been found between LUAD and LUSC samples. To further investigate autophagy in lung cancers, we focused our analysis on LUAD, which is holding the predominant position among all the pathological types of lung cancer. Performing differential expression analysis of 232 autophagy-related genes between tumor and adjacent tissue samples, we observed two clusters of patients according to the expression of autophagy signature A (23 genes) and signature B (12 genes). Patients in cluster 1, characterized by lower expression of signature A and higher expression of signature B than cluster 2, had the worst overall survival (OS) and disease-specific survival (DSS). Univariate and multivariate COX analyses suggested that autophagy signatures could be an independent feature associated with bad prognosis in patients. We showed that cluster 1 was more metabolically active, expressing anabolism-related genes involved in cell proliferation and migration. On the contrary, cluster 2 exhibited an antiproliferative phenotype, with active catabolism pathways. In addition, cluster 1 samples were less infiltrated by immune cells than cluster 2 and exhibited decreased immune response features. Analyses of autophagy signatures expression in single cells or sorted-cell datasets revealed that signature B was largely expressed by malignant cells, while signature A was preferentially expressed in endothelial and in less extend in fibroblast cells. Signature B expression in malignant cells correlated with an active metabolic feature, a decrease in immune cell infiltration and an increase in the immune checkpoint expression on tumor cells (e.g., PD-L1 and Galectin-9).
Interestingly, signature B correlated with an active autophagy process, supporting the central role for autophagy in tumor proliferation and migration and suggesting an important impact of this process on immune escape.
Previous studies analyzed the expression of autophagy genes in lung tumors and constructed the autophagy-related signature to anticipate the prognosis of LUAD or LUSC patients using the TCGA datasets [
15,
16,
22]. Two studies also determined predictive signatures based on autophagy-associated long non-coding RNAs [
14,
23]. The goal of this study was not to determine a new predictive signature based on autophagy-related genes. Instead, we investigated the expression of autophagy genes to understand the involvement of this process in lung tumor homeostasis. While previous studies established autophagy signatures in the global TME and correlated the expression of autophagy genes with the survival probability, we focused our analysis on malignant cells, highlighting a new autophagy signature that could help to understand the metabolic and immunologic status of these cells.
We take advantage of sorted cells and single cell datasets to analyze the expression of our signature and showed that signature B correlated with active metabolic status of tumor cells. Interestingly, the patients with high expression of signature B expressed much higher expression of autophagy genes involved in core machinery, including genes involved in the initiation complex (e.g.,
BECN1,
ULK1,
AMBA1) or elongation system (e.g., the majority of ATG genes,
MAP1LC3A,
MAP1LC3B,
MAP1LC3C). These data supported previous studies that described a pro-tumor function for autophagy in lung cancers, favoring the proliferation and migration of tumor cells [
3,
20,
23,
24].
Our data also revealed an important impact of autophagy on immune escape, describing that autophagy gene expression can reflect the immune cell infiltration and/or the immunogenic status of malignant cells. Interestingly, the correlation between autophagy genes expression and immune infiltration has also been described in other types of tumors [
23,
25]. While some studies observed a similar correlation between autophagy and immune checkpoint expression [
24,
26], future research needs to be developed to carefully understand the impact of autophagy in this context.
Among the genes involved in signature B, some of them reveal a significant prognostic value in TCGA cohorts. We showed that
ERO1L gene was preferentially expressed by malignant cells, suggesting an important role of this protein in the growth of cancer cells.
ERO1L has already been demonstrated to play a critical role in NSCLC, promoting cancer development by modulating cell cycle-related molecules [
27]. Moreover, recent reports also mentioned that ERO1L was implicated in anti-tumor immune response, by preventing T cell-mediated immunity and favoring myeloid suppressor cell activation [
28,
29]. The expression of
ERO1L gene in malignant cells could explain, at least in part, the reason for which signature B is associated with a bad prognosis, low infiltration of immune cells and high proliferative rates. Similarly,
ATIC was much more highly expressed in malignant cells and was associated with a significant prognostic value in our univariate analysis. A recent report demonstrated that ATIC facilitates tumor growth and migration by upregulating Myc expression in LUAD [
30].
BIRC5, an ATG12-ATG5 conjugate interactor has also been found to be expressed predominantly by tumor cells. BIRC5 was associated with a bad prognosis in lung cancers by favoring mitotic cell cycle-related pathways [
31]. In some tumors, BIRC5 was also correlated with high immune cells infiltration [
31]. In LUAD,
BIRC5 gene was inversely correlated with dendritic cells and CD4+ T cell infiltration, observations that we confirmed in our analysis. In our signature A, only
DRAM1 was preferentially expressed by tumor cells. DRAM1 was associated with p53 and played a critical role in autophagy and apoptosis [
32]. However, the biological function of DRAM1 in lung cancer remains controversial. The study by He Q et al. revealed that DRAM1 could be a target of FTSJ1 and promotes cancer progression [
33]. More recently, another study showed that DRAM1 inhibits the development of lung tumors by promoting the lysosomal degradation of EGFR [
34]. In our analysis, we showed that
DRAM1 was associated with a good prognostic value, suggesting that the expression of DRAM1 in malignant cells could inhibit tumor growth. Our data highlighted the vital role of these genes expressed by malignant cells in tumor development. While our study strongly suggests a correlation between the expression of these genes and the autophagy level in cancer cells, future investigations should be initiated to understand their role in autophagy modulation in the context of tumor growth.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}