
Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
- –
- On the discovery of cholestasis-related genes;
- –
- On pathological pathways of mutations in these loci;
- –
- On epidemiology and clinical features of patients with hereditary cholestatic diseases, focusing on the dysregulation of BAs in the liver gut axis concerning the development of primary biliary and liver cancers.
- –
- Abstracts or posters of congresses and meetings;
- –
- Editorials;
- –
- Studies that included patients with intrahepatic cholestasis without a genetic or histological diagnosis;
- –
- Articles focused on patients with other causes of autoimmune or acquired cholestasis, increased BAs such as primary biliary cholangitis, primary sclerosing cholangitis, IgG4 cholangitis, alcoholic and nonalcoholic steatohepatitis.
3. From Rare Pediatric Cholestatic Diseases to Adult Cryptogenic Cholestasis: An Overview of Different Features of Familial Intrahepatic Cholestasis
- –
- ATP8B1 gene (PFIC1): it is responsible for the synthesis of a lipid flippase, able to maintain the asymmetry of the cell membrane by the translocation of phospholipids from the exoplasmic to the cytoplasmic leaflet, having a protective role against excessive concentrations of BAs [14];
- –
- ABCB11 gene (PFIC2): coding for the bile export pump (BSEP), ABCB11 regulates the excretion of monovalent BAs from hepatocytes to bile canaliculi against a concentration gradient. The accumulation of BAs in hepatocytes is induced by a less expression or a malfunction of BSEP, resulting in cellular injury and alterations of the enterohepatic pathway of BAs [14];
- –
- ABCB4 gene (PFIC3): alterations of the MDR3 glycoprotein, a phosphatidylcholine flippase sited in the canalicular membrane of hepatocytes, lead to the disease; MDR3 protein carries phosphatidylcholine from the hepatocytes into the bile canaliculus, protecting the cholangiocytes from the detergent activity of BAs and reducing cellular injury. Patients with PFIC3 have, in fact, late onset of disease, and present high levels of gamma-glutamyl transferase (GGT) compared to PFIC1 and PFIC2, in which GGT is low or in the normal range [14];
- –
- TJP2 gene (PFIC4): TJP2 encodes an essential protein in the structure of tight junctions, establishing connections between the transmembrane tight junction proteins and the actin cytoskeleton. PFIC4 usually affects pediatric patients with low GGT levels [12];
- –
- NR1H4 gene (PFIC5): NR1H4 produces the farnesoid x receptor (FXR), bile acid-activated nuclear hormone receptor, the primary regulator of BAs metabolism and homeostasis [14];
- –
- SLC51A (PFIC6): this gene is responsible for the synthesis of the alpha subunit of the alpha-beta heteromeric organic solute transporter (OSTα-OSTβ), having the central role in the intestinal BA reabsorption in the setting of enterohepatic circulation: the pump exports BA across the basolateral membrane and OSTα deficiency causes a pediatric clinical picture with elevated liver transaminases, high GGT-cholestasis, normal serum BAs and congenital diarrhoea [15,16];
- –
- USP53 (PFIC7): USP53 encodes a nonprotease homolog of the ubiquitin-specific peptidase family; mutations in this gene are responsible for an autosomal recessive liver disorder characterized by infantile-onset jaundice and itching associated with cholestasis, elevated transaminases, normal GGT, hepatocellular and canalicular cholestasis with fibrotic changes at liver histology. In many cases, resolution of the liver injury is observed with age, although some patients have persistent hepatitis or splenomegaly. A subset of patients develops hearing loss since the USP53 locus interacts with the tight junction proteins TJP1 and TJP2 in polarized epithelial cells. Mutations in USP53 alter auditory hair cells modifying the stability of tight junctions [17];
- –
- KIF12 (PFIC8): mutations in KIF12 are characterized by cholestasis and high GGT presenting in the infantile period; liver immuno-staining of patients with KIF12 mutations resulted in changes in MRP2 (ABCC2 gene) trafficking with its strong cytoplasmic signal leading to change in cell polarity [18].
- –
- Benign recurrent intrahepatic cholestasis (BRIC): BRIC is an inherited disease characterized by almost two episodes of intermittent cholestasis with jaundice. Two types of BRIC are fully known, BRIC1 having mutations in the ATP8B1 gene and BRIC2 having mutations in the ABCB11 gene. The clinical presentation is usually less aggressive than PFIC since the protein function is only partially injured; pregnancy, infections, or drugs can trigger the attacks, while liver tests are normal between two episodes [19]. In the last years also, defects in the MYO5B gene have been described in patients with recurrent and transient forms of intrahepatic cholestasis [20,21]. Bull et al. reported BRIC-like phenotypes in a patient with USP53 mutations: age-onset was between infancy and 15 years, and patients had recurrent attacks of cholestasis with low GGT, hyperbilirubinemia and variably increased transaminases [22].
- –
- Intrahepatic cholestasis of pregnancy (ICP): the disease is the most common liver disorder of pregnancy, historically ascribed to the heterozygous mutations in ATP8B1, ABCB11 and ABCB4 genes. There are also associations with NR1H4 e TJP2 genes [23,24]. The main features are transient cholestasis and itching during the pregnancy that resolve after childbirth. Serious fetal complications are rare and occur when levels of BAs are higher than 40 µmol/L with ursodeoxycholic acid (UDCA) as the first-line therapy.
- –
- Low-phospholipid-associated cholelithiasis (LPAC): gallstone disease is linked to mutations in the ABCB4 gene in young people (younger than 40 years old) with symptomatic intrahepatic biliary lithiasis before and later even after cholecystectomy. A worsened secretion of phospholipids in the bile decreases the solubility of cholesterol, promoting gallstone formation. For this reason, long-term therapy with UDCA prevents several complications, especially after cholecystectomy, such as secondary sclerosing cholangitis [27].
4. Progressive Familial Intrahepatic Cholestasis-Related Genes
4.1. Role of Next-Generation Sequencing
4.2. Hepatobiliary Cancers and Cholestasis-Related Genes: An Underestimated Association
4.3. Hepatobiliary and Nor Cancers in Patients with Mutations in Cholestasis-Related Genes: PFIC Genes History and Cholestasis-Cancer Links
4.3.1. ATP8B1
4.3.2. ABCB11
- –
- BSEP1, if carrying at least one of the two common European mutations associated with residual BSEP function (c.1445A>G or c.890A>G);
- –
- BSEP2, if carrying at least one missense mutation, different to (c.1445A>G or c.890A>G);
- –
- BSEP3, if carrying mutations causing non-functional protein.
4.3.3. ABCB4
4.3.4. TJP2
- –
- Nine patients required LT;
- –
- One child died at 13 months;
- –
- Two had stable liver disease with mild portal hypertension at the ages of 4 and 7 years.
4.3.5. NR1H4
- –
- The increase in the synthesis of fibroblast growth factor-19 (FGF-19);
- –
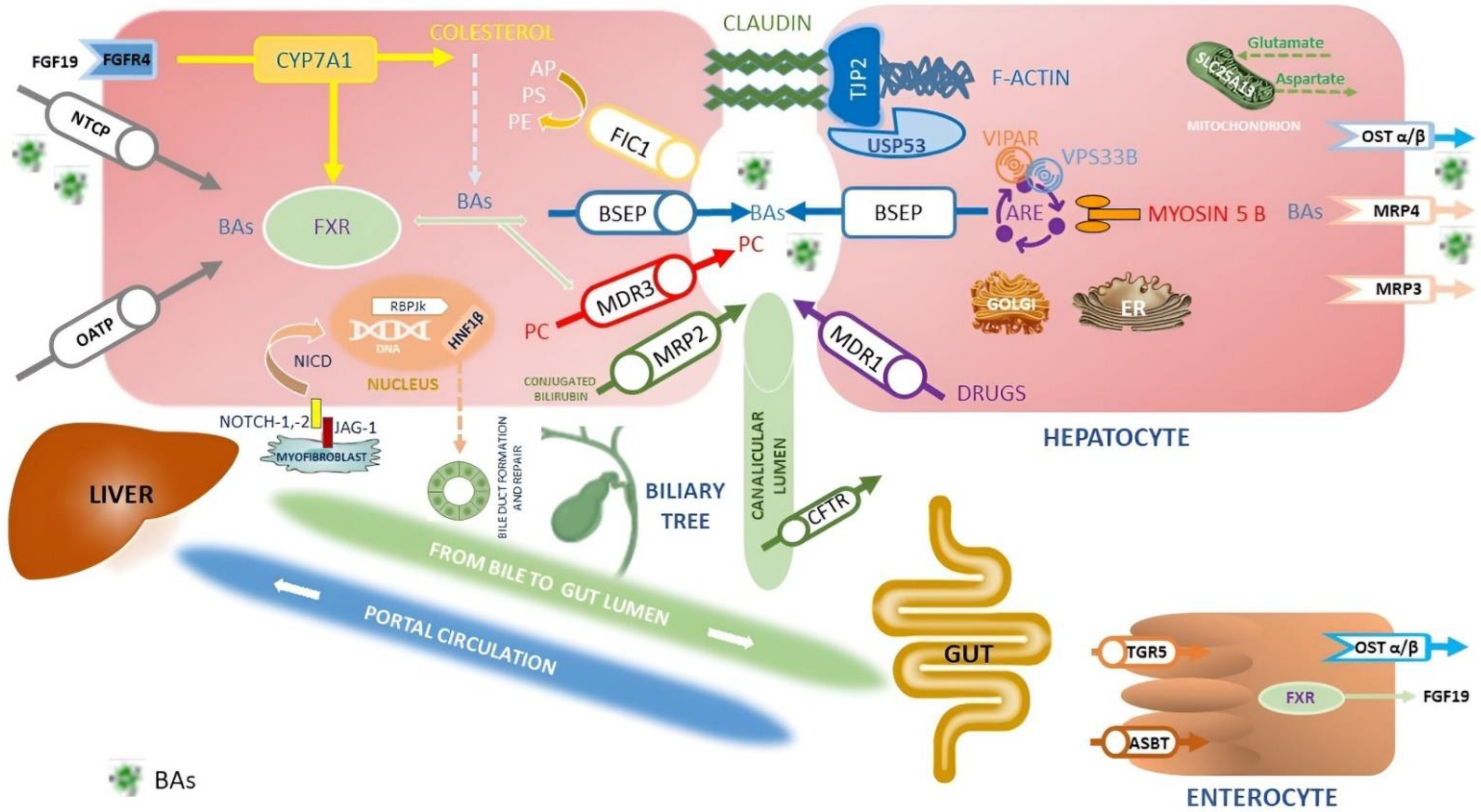
- The CY7A1 inhibition through the fibroblast growth factor receptor 4 (FGFR4) pathway in the hepatocytes (Figure 1);
- –
- The downregulation of sodium taurocholate cotransporting polypeptide (NTCP) blocking the uptake of BAs by the liver;
- –
- The upregulation of the synthesis of BSEP and MDR3;
- –
- The increase of BAs efflux from the liver to the lumen of bile canaliculus;
- –
- The synthesis of organic solute transporter alpha/beta (OSTα/β) via enhancement of BAs output from the liver to the portal vein [80].
4.3.6. MYO5B
4.3.7. SLC51A
4.3.8. USP53
- –
- In clear cell renal cell carcinoma inhibits the occurrence and development of cancer through NF-κB pathway inactivation [93];
- –
- In cervical squamous cell carcinoma correlated with the sensitivity to radiotherapy [94];
- –
- In oesophagal carcinoma, USP53 suppresses cancer progression by regulating cell growth and metabolism [95];
- –
- In lung adenocarcinoma, USP53 regulates cell apoptosis and glycolysis through the AKT1 pathway acting as a tumour suppressor [96].
4.3.9. KIF12
4.3.10. SLC25A13
4.3.11. JAG1 and NOTCH2
4.3.12. HNF1B
4.4. Other DNA Changes
5. Bile Acids and Liver Cancer: Pathophysiology
5.1. Structural and Functional Hepatocyte Polarity and Liver Disease
- –
- Claudin 1 (NISCH syndrome) and TJP2 (involved in PFIC4 and familial hypercholanaemia) in the group of tight junction proteins;
- –
- VPS33B (ARC syndrome) and Myosin 5B (involved in MVID and PFIC) in the intracellular trafficking protein;
- –
- ATP8B1 (PFIC1), ABCB11 (PFIC2), ABCB4 (PFIC3), and ABCC2 (involved in Dubin Johnson syndrome) in the canalicular membrane transporters;
- –
- SLCO1B1 and SLCO1B3 among the basolateral membrane transporters (Rotor syndrome), localized to the sinusoidal membrane of hepatocytes and able to control sodium-independent cellular uptake of bilirubin glucuronide, BAs, steroids and drugs;
- –
- –
- KIF12 is able to impair the hepatocyte polarity by interacting with MRP2 protein (ABCC2 gene), which has a strong cytoplasmic signal, in PFIC8-affected liver [17].
5.2. The Gut Microbiome–Bile Acid Axis in Hepatocarcinogenesis
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Arabic |
| AFP | alpha-phetoprotein |
| ARE | apical recycling endosome |
| ASBT | apical sodium-dependent bile acid transporter |
| AP | amino-phospholipids |
| BAs | bile acids |
| BD | biliary diversion |
| BSEP | bile salt export pump protein |
| BRIC | benign recurrent intrahepatic cholestasis |
| C | Caucasian |
| CA | Central Asian |
| CAC | Central Asian Caucasian |
| CCA | cholangiocarcinoma |
| Ch | Chinese |
| Cho | cholestasis |
| Cir | cirrhosis |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CNVs | copy number variants |
| CTLDN2 | adult-onset type II citrullinemia |
| CYP7A1 | cholesterol 7α-monooxygenase |
| del | deletion |
| DIC | drug-induced cholestasis |
| DILI | drug-induced liver injury |
| DSBs | double-strand breaks |
| ER | endoplasmic reticulum |
| FGF19 | fibroblast growth factor 19 |
| FGFR14 | fibroblast growth factor receptor 4 |
| FIC | familial intrahepatic cholestasis |
| FIC 1 | familial intrahepatic cholestasis deficiency type 1 protein |
| FXR | farnesoid X receptor |
| FTTDCD | failure to thrive and dyslipidemia caused by citrin deficiency |
| GGT | gamma-glutamyl transferase |
| GWAS | genome-wide association study |
| H | hepatomegaly |
| Hi | Hispanic |
| HBCs | hepatobiliary cancers |
| HCC | hepatocellular carcinoma |
| HNF-1B | hepatocyte nuclear factor-1beta |
| I | itching |
| IBAT | ileal bile acid transporter |
| ICP | intrahepatic cholestasis of pregnancy |
| INDELs | DNA insertions and deletions |
| J | jaundice |
| Ja | Japanese |
| JAG-2 | jagged canonical notch ligand-2 |
| KIF12 | kinesin family member 12 |
| LF | liver failure |
| LT | liver transplantation |
| MODY | maturity-onset diabetes of the young |
| MVID | microvillus inclusion disease |
| LPAC | low-phospholipid-associated cholelithiasis |
| LT | liver transplantation |
| MB | megabases |
| MDR | multidrug resistance protein |
| mo | months |
| MRP | multidrug resistance protein |
| NEC | Northern European Caucasian |
| NGS | next-generation sequencing |
| mo | months |
| MDR | multidrug resistance protein |
| ng | nanograms/mL |
| NKT | natural killer T |
| nf | not found |
| NICD | notch intracellular domain |
| NLT | normalized liver tests |
| NOTCH-1,2 | notch homolog-1,2 translocation-associated |
| NTCP | sodium taurocholate cotransporting polypeptide |
| OMIM | Online Mendelian Inheritance in Man |
| OR | odd ratio |
| OATP | organic anion transporting polypeptide |
| OST α/β | organic solute transporter alpha/beta |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PELD PFIC | paediatric end-stage liver disease progressive familial intrahepatic cholestasis |
| PGS | panel gene sequencing |
| PMID | PubMed-Indexed for MEDLINE |
| PS | phosphatidylserine |
| RBPjk | recombining binding protein suppressor of hairless |
| RCAD | renal cysts and diabetes syndrome |
| ROS | reactive oxygen species |
| SA | South Asian |
| SL25A13 | solute carrier family 25 member 13 |
| STRs | short tandem repeats |
| SVs | structural variants |
| T | Taiwanese |
| TGR5 | G-protein-coupled bile acid receptor |
| TJP2 | tight junction protein 2 gene |
| THBA | tetrahydroxylated bile acids |
| UDCA | ursodeoxycholic acid |
| USP53 | ubiquitin-specific peptidase 53 |
| VIPAR | VPS33B interacting protein, apical–basal polarity regulator |
| VPS33B | vacuolar protein sorting associated protein 33B |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
| wk | weeks |
| wt | wild type |
| y | year |
| yrs | years |
References
- Vitale, G.; Gitto, S.; Raimondi, F.; Mattiaccio, A.; Mantovani, V.; Vukotic, R.; D’Errico, A.; Seri, M.; Russell, R.B.; Andreone, P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J. Gastroenterol. 2018, 53, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Feng, J.; Li, J.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed. Pharm. 2020, 133, 111036. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Jeon, C.O. Promotion and induction of liver cancer by gut microbiome-mediated modulation of bile acids. PLoS Pathog. 2019, 15, e1007954. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Kamath, B.M.; Loomes, K.M.; Karpen, S.J. Cholestatic liver diseases of genetic etiology: Advances and controversies. Hepatology 2022, 75, 1627–1646. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511. [Google Scholar] [CrossRef]
- Goldberg, A.; Mack, C.L. Inherited Cholestatic Diseases in the Era of Personalized Medicine. Clin. Liver Dis. 2020, 15, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Nayagam, J.S.; Williamson, C.; Joshi, D.; Thompson, R.J. Review Article: Liver Disease in Adults with Variants in the Cho-lestasis-Related Genes ABCB11, ABCB4 and ATP8B1. Aliment. Pharmacol. Ther. 2020, 52, 1628–1639. [Google Scholar] [CrossRef]
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.M. Primary malignant tumours in the non-cirrhotic liver. Eur. J. Radiol. 2017, 95, 349–361. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Kriegermeier, A.; Green, R. Pediatric Cholestatic Liver Disease: Review of Bile Acid Metabolism and Discussion of Current and Emerging Therapies. Front. Med. 2020, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sun, Y.; van Ijzendoorn, S.C.D. A Link between Intrahepatic Cholestasis and Genetic Variations in Intracellular Trafficking Regulators. Biology 2021, 10, 119. [Google Scholar] [CrossRef]
- Bull, L.N.; Thompson, R. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Gao, E.; Cheema, H.; Waheed, N.; Mushtaq, I.; Erden, N.; Nelson-Williams, C.; Jain, D.; Soroka, C.J.; Boyer, J.L.; Khalil, Y.; et al. Organic Solute Transporter Alpha Deficiency: A Disorder With Cholestasis, Liver Fibrosis, and Congenital Diarrhea. Hepatology 2020, 71, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Rao, A.; Elpeleg, O.; Vaz, F.M.; Abu Libdeh, B.Y.; Karpen, S.J.; Dawson, P.A. Organic solute transporter-β (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology 2018, 68, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- Stalke, A.; Sgodda, M.; Cantz, T.; Skawran, B.; Lainka, E.; Hartleben, B.; Baumann, U.; Pfister, E.-D. KIF12 Variants and Disturbed Hepatocyte Polarity in Children with a Phenotypic Spectrum of Cholestatic Liver Disease. J. Pediatr. 2022, 240, 284–291.e9. [Google Scholar] [CrossRef]
- Vitale, G.; Gitto, S.; Vukotic, R.; Raimondi, F.; Andreone, P. Familial intrahepatic cholestasis: New and wide perspectives. Dig. Liver Dis. 2019, 51, 922–933. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Gong, J.-Y.; Feng, J.-Y.; Wang, R.-X.; Han, J.; Liu, T.; Lu, Y.; Li, L.-T.; Zhang, M.-H.; Sheps, J.A.; et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis. Hepatology 2017, 65, 1655–1669. [Google Scholar] [CrossRef]
- Wang, L.; Qiu, Y.-L.; Xu, H.-M.; Zhu, J.; Li, S.-J.; OuYang, W.-X.; Yang, Y.-F.; Lu, Y.; Xie, X.-B.; Xing, Q.-H.; et al. MYO5B-associated diseases: Novel liver-related variants and genotype-phenotype correlation. Liver Int. 2022, 42, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Ellmers, R.; Foskett, P.; Strautnieks, S.; Sambrotta, M.; Czubkowski, P.; Jankowska, I.; Wagner, B.; Deheragoda, M.; Thompson, R.J. Cholestasis Due to USP53 Deficiency. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Van Mil, S.W.C.; Milona, A.; Dixon, P.H.; Mullenbach, R.; Geenes, V.L.; Chambers, J.; Shevchuk, V.; Moore, G.E.; Lammert, F.; Glantz, A.G.; et al. Functional Variants of the Central Bile Acid Sensor FXR Identified in Intrahepatic Cholestasis of Pregnancy. Gastroenterology 2007, 133, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P.H.; Sambrotta, M.; Chambers, J.; Taylor-Harris, P.; Syngelaki, A.; Nicolaides, K.; Knisely, A.S.; Thompson, R.J.; Williamson, C. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 2017, 7, 11823. [Google Scholar] [CrossRef] [Green Version]
- Wikström Shemer, E.A.; Stephansson, O.; Thuresson, M.; Thorsell, M.; Ludvigsson, J.F.; Marschall, H.-U. Intrahepatic cholestasis of pregnancy and cancer, immune-mediated and cardiovascular diseases: A population-based cohort study. J. Hepatol. 2015, 63, 456–461. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Serrano, J.; Knoben, J.E.; Navarro, V.J. LiverTox: A website on drug-induced liver injury. Hepatology 2012, 57, 873–874. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Poupon, R. Low phospholipid associated cholelithiasis: Association with mutation in the MDR3/ABCB4 gene. Orphanet J. Rare Dis. 2007, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2007, 5, 16–18. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. Am. J. Hum. Genet. 2007, 80, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, A.P.; Zhu, D.; Kidambi, A.; Ly, C.; Tey, S.; Brewer, M.H.; Ahmad-Annuar, A.; Nicholson, G.A.; Kennerson, M.L. Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Evans, J.M.; Olson, T.M. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am. J. Med Genet. Part A 2015, 167, 886–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X. Exome sequencing greatly expedites the progressive research of Mendelian diseases. Front. Med. 2014, 8, 42–57. [Google Scholar] [CrossRef]
- Rexach, J.; Lee, H.; Martinez-Agosto, J.A.; Németh, A.H.; Fogel, B.L. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019, 18, 492–503. [Google Scholar] [CrossRef]
- Guo, Y.; Long, J.; He, J.; Li, C.-I.; Cai, Q.; Shu, X.-O.; Zheng, W.; Li, C. Exome sequencing generates high quality data in non-target regions. BMC Genom. 2012, 13, 194. [Google Scholar] [CrossRef] [Green Version]
- Brar, T.S.; Hilgenfeldt, E.; Soldevila-Pico, C. Etiology and Pathogenesis of Hepatocellular Carcinoma. In Precision Molecular Pathology of Liver Cancer; Liu, C., Ed.; Molecular Pathology Library; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–15. ISBN 978-3-319-68082-8. [Google Scholar]
- Knisely, A.S.; Strautnieks, S.S.; Meier, Y.; Stieger, B.; Byrne, J.A.; Portmann, B.C.; Bull, L.N.; Pawlikowska, L.; Bilezikçi, B.; Özçay, F.; et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006, 44, 478–486. [Google Scholar] [CrossRef]
- Davit-Spraul, A.; Fabre, M.; Branchereau, S.; Baussan, C.; Gonzales, E.; Stieger, B.; Bernard, O.; Jacquemin, E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010, 51, 1645–1655. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Strautnieks, S.; Jankowska, I.; Czubkowski, P.; Emerick, K.; Antoniou, A.; Wanty, C.; Fischler, B.; Jacquemin, E.; Wali, S.; et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J. Hepatol. 2010, 53, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Hertel, P.M.; Finegold, M.J.; Wang, L.; Kerkar, N.; Wang, J.; Wong, L.C.; Plon, S.E.; Sambrotta, M.; Foskett, P.; et al. Hepatocellular carcinoma associated with tight-junction protein 2 deficiency. Hepatology 2015, 62, 1914–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrotta, M.; Strautnieks, S.; Papouli, E.; Rushton, P.; Clark, B.E.; Parry, D.A.; Logan, C.V.; Newbury, L.J.; Ka-math, B.M.; Ling, S.; et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat. Genet. 2014, 46, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vij, M.; Shanmugam, N.P.; Reddy, M.S.; Govil, S.; Rela, M. Hepatocarcinogenesis in multidrug-resistant P-glycoprotein 3 deficiency. Pediatr. Transpl. 2017, 21, e12889. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddsson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldórsson, B.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444. [Google Scholar] [CrossRef]
- Lammert, F.; Hochrath, K. A letter on ABCB4 from Iceland: On the highway to liver disease. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Collino, A.; Sinha, S.K.; Radaelli, E.; Nicoli, P.; D’Antiga, L.; Sonzogni, A.; Faivre, J.; Buendia, M.A.; Sturm, E.; et al. Massive gene amplification drives paediatric hepatocellular carcinoma caused by bile salt export pump deficiency. Nat. Commun. 2014, 5, 3850. [Google Scholar] [CrossRef]
- Jüngst, C.; Justinger, C.; Fischer, J.; Berg, T.; Lammert, F. Common ABCB4 and ABCB11 Genotypes Are Associated with Idiopathic Chronic Cholestasis in Adults. Dig. Dis. 2021, 40, 489–496. [Google Scholar] [CrossRef]
- Clayton, R.J.; Iber, F.L.; Ruebner, B.H.; McKusick, V.A. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. Am. J. Dis. Child. 1969, 117, 112–124. [Google Scholar] [CrossRef]
- De Vos, R.; de Wolf-Peeters, C.; Desmet, V.; Eggermont, E.; Van Acker, K. Progressive intrahepatic cholestasis (Byler’s disease): Case report. Gut 1975, 16, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, I.-M.; Ørnvold, K.; Jacobsen, B.B.; Ranek, L. Fatal Familial Cholestatic Syndrome in Greenland Eskimo Children. Acta Paediatr. Scand. 1986, 75, 1010–1016. [Google Scholar] [CrossRef]
- Jacquemin, E.; Dumont, M.; Bernard, O.; Erlinger, S.; Hadchouel, M. Evidence for defective primary bile acid secretion in children with progressive familial intrahepatic cholestasis (Byler disease). Eur. J. Pediatr. 1994, 153, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Carlton, V.E.; Knisely, A.; Freimer, N.B. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum. Mol. Genet. 1995, 4, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; van Eijk, M.J.; Pawlikowska, L.; DeYoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.; Lomri, N.; Berger, R.; Scharschmidt, B.F.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 1998, 18, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sándor, T.; Surinya, M.; Mónus, Z. Familial occurrence of giant cell hepatitis in infancy. Acta Hepatogastroenterol. 1976, 23, 101–104. [Google Scholar]
- Strautnieks, S.S.; Bull, L.N.; Knisely, A.S.; Kocoshis, S.A.; Dahl, N.; Arnell, H.; Sokal, E.; Dahan, K.; Childs, S.; Ling, V.; et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat. Genet. 1998, 20, 233–238. [Google Scholar] [CrossRef]
- Noe, J.; Kullak-Ublick, G.A.; Jochum, W.; Stieger, B.; Kerb, R.; Haberl, M.; Mullhaupt, B.; Meier, P.J.; Pauli-Magnus, C. Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J. Hepatol. 2005, 43, 536–543. [Google Scholar] [CrossRef]
- Hayashi, H.; Takada, T.; Suzuki, H.; Akita, H.; Sugiyama, Y. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology 2005, 41, 916–924. [Google Scholar] [CrossRef]
- Van Mil, S.W.C.; van der Woerd, W.L.; van der Brugge, G.; Sturm, E.; Jansen, P.L.M.; Bull, L.N.; van den Berg, I.E.T.; Berger, R.; Houwen, R.H.J.; Klomp, L.W.J. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 2004, 127, 379–384. [Google Scholar] [CrossRef]
- Leabman, M.K.; Huang, C.C.; DeYoung, J.; Carlson, E.J.; Taylor, T.R.; de la Cruz, M.; Johns, S.J.; Stryke, D.; Kawamoto, M.; Urban, T.J.; et al. Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc. Natl. Acad. Sci. USA 2003, 100, 5896–5901. [Google Scholar] [CrossRef] [Green Version]
- Scheimann, A.O.; Strautnieks, S.S.; Knisely, A.S.; Byrne, J.A.; Thompson, R.J.; Finegold, M.J. Mutations in Bile Salt Export Pump (ABCB11) in Two Children with Progressive Familial Intrahepatic Cholestasis and Cholangiocarcinoma. J. Pediatr. 2007, 150, 556–559. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; Lang, T.; Meier, Y.; Zodan-Marin, T.; Jung, D.; Breymann, C.; Zimmermann, R.; Kenngott, S.; Beuers, U.; Reichel, C.; et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics 2004, 14, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, C.; Meier, Y.; Stieger, B.; Beuers, U.; Lang, T.; Kerb, R.; Kullak-Ublick, G.A.; Meier, P.J.; Pauli-Magnus, C. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharm. Genom. 2007, 17, 47–60. [Google Scholar] [CrossRef] [PubMed]
- AlSalloom, A. Hepatocellular Carcinoma in a Boy with Progressive Familial Intrahepatic Cholestasis Type II: Challenging Identification: Case Report. Int. J. Health Sci. 2013, 7, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, S.; Erson-Omay, E.Z.; Harmanci, A.S.; Morotti, R.; Carrion-Grant, G.; Baranoski, J.; Knisely, A.S.; Ekong, U.; Emre, S.; Yasuno, K.; et al. Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J. Hepatol. 2014, 61, 1178–1183. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe Bile Salt Export Pump Deficiency: 82 Different ABCB11 Mutations in 109 Families. Gastroenterology 2008, 134, 1203–1214.e8. [Google Scholar] [CrossRef] [Green Version]
- Van Wessel, D.B.E.; Thompson, R.J.; Gonzales, E.; Jankowska, I.; Sokal, E.; Grammatikopoulos, T.; Kadaristiana, A.; Jacquemin, E.; Spraul, A.; Lipiński, P.; et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J. Hepatol. 2020, 73, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Deleuze, J.F.; Jacquemin, E.; Dubuisson, C.; Cresteil, D.; Dumont, M.; Erlinger, S.; Bernard, O.; Hadchouel, M. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 1996, 23, 904–908. [Google Scholar] [CrossRef]
- De Vree, J.M.; Jacquemin, E.; Sturm, E.; Cresteil, D.; Bosma, P.J.; Aten, J.; Deleuze, J.-F.; Desrochers, M.; Burdelski, M.; Bernard, O.; et al. Mutations in the MDR 3 gene cause progressive familial intrahepatic cholestasis. Proc. Natl. Acad. Sci. USA 1998, 95, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Wendum, D.; Barbu, V.; Rosmorduc, O.; Arrivé, L.; Fléjou, J.-F.; Poupon, R. Aspects of liver pathology in adult patients with MDR3/ABCB4 gene mutations. Virchows Arch. 2012, 460, 291–298. [Google Scholar] [CrossRef]
- Poupon, R.; Rosmorduc, O.; Boëlle, P.Y.; Chrétien, Y.; Corpechot, C.; Chazouillères, O.; Housset, C.; Barbu, V. Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: A study of 156 consecutive patients. Hepatology 2013, 58, 1105–1110. [Google Scholar] [CrossRef]
- Vij, M.; Safwan, M.; Shanmugam, N.P.; Rela, M. Liver pathology in severe multidrug resistant 3 protein deficiency: A series of 10 pediatric cases. Ann. Diagn. Pathol. 2015, 19, 277–282. [Google Scholar] [CrossRef] [PubMed]
- de Vries, E.; Mazzetti, M.; Takkenberg, B.; Mostafavi, N.; Bikker, H.; Marzioni, M.; de Veer, R.; van der Meer, A.; Doukas, M.; Verheij, J.; et al. Carriers of ABCB4 gene variants show a mild clinical course, but impaired quality of life and limited risk for cholangiocarcinoma. Liver Int. 2020, 40, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Shanmugam, N.P.; Reddy, M.S.; Sankaranarayanan, S.; Rela, M. Paediatric hepatocellular carcinoma in tight junction protein 2 (TJP2) deficiency. Virchows Arch. 2017, 471, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-S.; Becher, N.; Friis, J.B.; Ott, P.; Vogel, I.; Grønbæk, H. New tight junction protein 2 variant causing progressive familial intrahepatic cholestasis type 4 in adults: A case report. World J. Gastroenterol. 2020, 26, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-Y.; Xiong, L.; Wray, C.G.; Ballatori, N.; Boyer, J.L. The Farnesoid X Receptor FXRalpha/NR1H4 Acquired Ligand Specificity for Bile Salts Late in Vertebrate Evolution. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1400–R1409. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Potter, C.J.; Xiao, R.; Manickam, K.; Kim, M.-S.; Kim, K.H.; Shneider, B.L.; Picarsic, J.L.; Ja-cobson, T.A.; Zhang, J.; et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat. Commun. 2016, 7, 10713. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Wang, L.-L.; Shan, Q.-W.; Tang, Q.; Deng, Y.-N.; Lian, S.-J.; Yun, X. A novel heterozygous NR1H4 termination codon mutation in idiopathic infantile cholestasis. World J. Pediatr. 2011, 8, 67–71. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef]
- Himes, R.W.; Mojarrad, M.; Eslahi, A.; Finegold, M.J.; Maroofian, R.; Moore, D.D. NR1H4-related Progressive Familial Intrahepatic Cholestasis 5: Further Evidence for Rapidly Progressive Liver Failure. J. Pediatr. Gastroenterol. Nutr. 2020, 70, e111–e113. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2006, 28, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariello, M.; Peres, C.; Zerlotin, R.; Porru, E.; Sabbà, C.; Roda, A.; Moschetta, A. Long-term Administration of Nuclear Bile Acid Receptor FXR Agonist Prevents Spontaneous Hepatocarcinogenesis in Abcb4−/− Mice. Sci. Rep. 2017, 7, 11203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Luo, Q.; Zeng, S.; Lou, Y.; Li, X.; Hu, M.; Lu, L.; Liu, Z. Disordered farnesoid X receptor signaling is associated with liver carcinogenesis in Abcb11 -deficient mice. J. Pathol. 2021, 255, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gong, W.; Dai, S.; Huang, G.; Shen, X.; Gao, M.; Xu, Z.; Zeng, Y.; He, F. Downregulation of Human Farnesoid X Receptor by miR-421 Promotes Proliferation and Migration of Hepatocellular Carcinoma Cells. Mol. Cancer Res. 2012, 10, 516–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.; Ma, L.; Tang, W.; Huang, P.; Yang, B.; Wang, L.; Chen, S.; Gao, Q.; Zhang, S.; Xia, J. FXR Acts as a Metastasis Suppressor in Intrahepatic Cholangiocarcinoma by Inhibiting IL-6-Induced Epithelial-Mesenchymal Transition. Cell. Physiol. Biochem. 2018, 48, 158–172. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef] [Green Version]
- Girard, M.; Lacaille, F.; Verkarre, V.; Mategot, R.; Feldmann, G.; Grodet, A.; Sauvat, F.; Irtan, S.; Davit-Spraul, A.; Jacquemin, E.; et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 2014, 60, 301–310. [Google Scholar] [CrossRef]
- Gonzales, E.; Taylor, S.A.; Davit-Spraul, A.; Thébaut, A.; Thomassin, N.; Guettier, C.; Whitington, P.F.; Jacquemin, E. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2017, 65, 164–173. [Google Scholar] [CrossRef]
- Kuang, S.-Q.; Tong, W.-G.; Yang, H.; Lin, W.; Lee, M.K.; Fang, Z.H.; Wei, Y.; Jelinek, J.; Issa, J.-P.; Garcia-Manero, G. Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia 2008, 22, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Chen, X.; Chen, P.; Yue, D.; Zhu, L.; Fan, Q. Inactivation of MYO5B Promotes Invasion and Motility in Gastric Cancer Cells. Am. J. Dig. Dis. 2012, 57, 1247–1252. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Gong, J.-Y.; Li, L.-T.; Li, J.-Q.; Zhang, M.-H.; Lu, Y.; Xie, X.-B.; Hong, Y.-R.; Yu, Z.; et al. Low-GGT Intrahepatic Cholestasis Associated with Biallelic USP53 Variants: Clinical, Histological and Ultrastructural Characterization. Liver Int. 2020, 40, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.; Dong, Z.; Peng, W.; Jiang, W.; Huang, G.; Liu, G.; Ye, Z.; Wang, Y.; Xu, Z.; Fu, J.; et al. Ubiquitin-specific peptidase 53 inhibits the occurrence and development of clear cell renal cell carcinoma through NF-κB pathway inactivation. Cancer Med. 2021, 10, 3674–3688. [Google Scholar] [CrossRef]
- Zhou, Q.; Yao, X.; Wu, C.; Chen, S.; Fan, D. Knockdown of Ubiquitin-Specific Protease 53 Enhances the Radiosensitivity of Human Cervical Squamous Cell Carcinoma by Regulating DNA Damage-Binding Protein 2. Technol. Cancer Res. Treat. 2020, 19, 1533033820929792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Tang, Y.; Tong, X.; Zhou, Q.; Xie, J.; Wang, J.; Han, Y.; Ta, N.; Ye, Z. USP53 activated by H3K27 acetylation regulates cell viability, apoptosis and metabolism in esophageal carcinoma via the AMPK signaling pathway. Carcinogenesis 2021, 43, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, X.; Wang, H.; Yu, H.; Wang, J. USP53 promotes apoptosis and inhibits glycolysis in lung adenocarcinoma through FKBP51-AKT1 signaling. Mol. Carcinog. 2020, 59, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Ünlüsoy Aksu, A.; Das, S.K.; Nelson-Williams, C.; Jain, D.; Özbay Hoşnut, F.; Evirgen Şahin, G.; Lifton, R.P.; Vi-larinho, S. Recessive Mutations in KIF12 Cause High Gamma-Glutamyltransferase Cholestasis. Hepatol. Commun. 2019, 3, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef] [PubMed]
- SSong, Y.-Z.; Deng, M.; Chen, F.-P.; Wen, F.; Guo, L.; Cao, S.-L.; Gong, J.; Xu, H.; Jiang, G.-Y.; Zhong, L.; et al. Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric center. Int. J. Mol. Med. 2011, 28, 33–40. [Google Scholar] [CrossRef]
- He, J.; Zhang, J.; Li, X.; Wang, H.; Feng, C.; Fang, F.; Shu, S. A Case Report: Can Citrin Deficiency Lead to Hepatocellular Carcinoma in Children? Front. Pediatr. 2019, 7, 371. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Yang, C.-C.; Chen, H.-L.; Hwu, W.-L.; Wu, M.-Z.; Liu, K.-L.; Wu, M.-S. Homozygous SLC25A13 Mutation in a Taiwanese Patient with Adult-onset Citrullinemia Complicated with Steatosis and Hepatocellular Carcinoma. J. Formos. Med. Assoc. 2006, 105, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Okamoto, Y.; Morita, T.; Matsumoto, M.; Fukui, H.; Nakano, H.; Tsujii, T. Promoting effect of citrulline in hepatocarconogenesis: Possbile mechanism in hypercitrullinemia. Hepatology 1990, 11, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Soeda, J.; Yazaki, M.; Nakata, T.; Miwa, S.; Ikeda, S.-I.; Hosoda, W.; Iijima, M.; Kobayashi, K.; Saheki, T.; Kojiro, M.; et al. Primary Liver Carcinoma Exhibiting Dual Hepatocellular-Biliary Epithelial Differentiations Associated With Citrin Deficiency: A Case Report. J. Clin. Gastroenterol. 2008, 42, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhu, S.; Zhang, M.; Dong, Y.; Wang, F.-S. A 6-Year-Old Child With Citrin Deficiency and Advanced Hepatocellular Carcinoma. Pediatrics 2018, 143, e20181931. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Krantz, I.D.; Deng, Y.; Genin, A.; Banta, A.B.; Collins, C.C.; Qi, M.; Trask, B.J.; Kuo, W.L.; Cochran, J.; et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997, 16, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, C.; Fornari, F.; Piscaglia, F.; Gramantieri, L. Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs. Cells 2021, 10, 521. [Google Scholar] [CrossRef]
- Fabris, L.; Cadamuro, M.; Guido, M.; Spirli, C.; Fiorotto, R.; Colledan, M.; Torre, G.; Alberti, D.; Sonzogni, A.; Okolicsanyi, L.; et al. Analysis of Liver Repair Mechanisms in Alagille Syndrome and Biliary Atresia Reveals a Role for Notch Signaling. Am. J. Pathol. 2007, 171, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Geisler, F.; Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 2015, 61, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morell, C.M.; Fiorotto, R.; Fabris, L.; Strazzabosco, M. Notch signalling beyond liver development: Emerging concepts in liver repair and oncogenesis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 447–454. [Google Scholar] [CrossRef]
- Békássy, A.N.; Garwicz, S.; Wiebe, T.; Hägerstrand, I.; Jensen, O.A. Hepatocellular carcinoma associated with arteriohepatic dysplasia in a 4-year-old girl. Med. Pediatr. Oncol. 1992, 20, 78–83. [Google Scholar] [CrossRef]
- Bhadri, V.A.; Stormon, M.O.; Arbuckle, S.; Lam, A.H.; Gaskin, K.J.; Shun, A. Hepatocellular Carcinoma in Children with Alagille Syndrome. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 676–678. [Google Scholar] [CrossRef]
- Chiaretti, A.; Zampino, G.; Botto, L.; Polidori, G. Alagille syndrome and hepatocarcinoma: A case report. Acta Paediatr. 1992, 81, 937. [Google Scholar] [CrossRef]
- Pérez Becerra, E.; Fuster, M.; Fraga, M.; Antúnez, J.; Pintos, E.; Pavón, P.; Forteza, J. Alagille’s syndrome: A family case and its association with hepatocellular carcinoma. Rev. Clin. Esp. 1991, 188, 459–462. [Google Scholar] [PubMed]
- Schwarzenberg, S.J.; Grothe, R.M.; Sharp, H.L.; Snover, D.C.; Freese, D. Long-term complications of arteriohepatic dysplasia. Am. J. Med. 1992, 93, 171–176. [Google Scholar] [CrossRef]
- Valamparampil, J.J.; Shanmugam, N.; Vij, M.; Reddy, M.S.; Rela, M. Hepatocellular Carcinoma in Paediatric Patients with Alagille Syndrome: Case Series and Review of Literature. J. Gastrointest. Cancer 2020, 51, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Schindler, E.A.; Gilbert, M.A.; Piccoli, D.A.; Spinner, N.B.; Krantz, I.D.; Loomes, K.M. Alagille syndrome and risk for hepatocellular carcinoma: Need for increased surveillance in adults with mild liver phenotypes. Am. J. Med. Genet. Part A 2021, 185, 719–731. [Google Scholar] [CrossRef]
- Kornfeld, J.-W.; Baitzel, C.; Könner, A.C.; Nicholls, H.T.; Vogt, M.C.; Herrmanns, K.; Scheja, L.; Haumaitre, C.; Wolf, A.M.; Knippschild, U.; et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature 2013, 494, 111–115. [Google Scholar] [CrossRef]
- Verdeguer, F.; Le Corre, S.; Fischer, E.; Callens, C.; Garbay, S.; Doyen, A.; Igarashi, P.; Terzi, F.; Pontoglio, M. A mitotic transcriptional switch in polycystic kidney disease. Nat. Med. 2010, 16, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Kotalova, R.; Dusatkova, P.; Cinek, O.; Dusatkova, L.; Dedic, T.; Seeman, T.; Lebl, J.; Pruhova, S. Hepatic phenotypes of HNF1B gene mutations: A case of neonatal cholestasis requiring portoenterostomy and literature review. World J. Gastroenterol. 2015, 21, 2550–2557. [Google Scholar] [CrossRef]
- De Leusse, C.; De Paula, A.M.; Ascherod, A.; Parache, C.; Hery, G.; Cailliez, M.; Missirian, C.; Fabre, A. Hepatocarcinoma and Cholestasis Associated to Germline Hemizygous Deletion of Gene HNF1B. J. Pediatr. Gastroenterol. Nutr. 2019, 68, e85. [Google Scholar] [CrossRef]
- Yu, D.-D.; Jing, Y.-Y.; Guo, S.-W.; Ye, F.; Lu, W.; Li, Q.; Dong, Y.-L.; Gao, L.; Yang, Y.-T.; Yang, Y.; et al. Overexpression Of Hepatocyte Nuclear Factor-1beta Predicting Poor Prognosis Is Associated With Biliary Phenotype In Patients With Hepatocellular Carcinoma. Sci. Rep. 2015, 5, srep13319. [Google Scholar] [CrossRef] [Green Version]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmierczak, M.; Harris, S.L.; Kazmierczak, P.; Shah, P.; Starovoytov, V.; Ohlemiller, K.K.; Schwander, M. Progressive Hearing Loss in Mice Carrying a Mutation in Usp53. J. Neurosci. 2015, 35, 15582–15598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of β-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Tanaka, N.; Fukami, T.; Xie, C.; Yagai, T.; Kim, D.; Velenosi, T.J.; Yan, T.; Krausz, K.W.; Levi, M.; et al. Role of Farnesoid X Receptor and Bile Acids in Hepatic Tumor Development. Hepatol. Commun. 2018, 2, 1567–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased Activation of the Wnt/β-Catenin Pathway in Spontaneous Hepatocellular Carcinoma Observed in Farnesoid X Receptor Knockout Mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Merlen, G.; Kahale, N.; Ursic-Bedoya, J.; Bidault-Jourdainne, V.; Simerabet, H.; Doignon, I.; Tanfin, Z.; Garcin, I.; Péan, N.; Gautherot, J.; et al. TGR5-dependent hepatoprotection through the regulation of biliary epithelium barrier function. Gut 2020, 69, 146–157. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2019, 68, 1014–1023. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Park, M.-Y.; Kim, S.J.; Ko, E.K.; Ahn, S.-H.; Seo, H.; Sung, M.-K. Gut microbiota-associated bile acid deconjugation accelerates hepatic steatosis in ob/ob mice. J. Appl. Microbiol. 2016, 121, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.W.F.; Dijk, W.; Boekhorst, J.; Kuipers, F.; Groen, A.K.; Lukovac, S.; Hooiveld, G.J.E.J.; Kersten, S. ANGPTL4 promotes bile acid absorption during taurocholic acid supplementation via a mechanism dependent on the gut microbiota. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Jena, P.K.; Hu, Y.; Liu, H.-X.; Nagar, N.; Kalanetra, K.M.; French, S.W.; French, S.W.; Mills, D.A.; Wan, Y.-J.Y. Hepatic inflammation caused by dysregulated bile acid synthesis is reversible by butyrate supplementation. J. Pathol. 2017, 243, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Yeoh, B.S.; Chassaing, B.; Xiao, X.; Saha, P.; Aguilera Olvera, R.; Lapek, J.D.; Zhang, L.; Wang, W.-B.; Hao, S.; et al. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell 2018, 175, 679–694.e22. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Sheps, J.A.; Wang, R.; Wang, J.; Ling, V. The protective role of hydrophilic tetrahydroxylated bile acids (THBA). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158925. [Google Scholar] [CrossRef]
- Wang, R.; Sheps, J.A.; Liu, L.; Han, J.; Chen, P.S.K.; Lamontagne, J.; Wilson, P.D.; Welch, I.D.; Borchers, C.H.; Ling, V. Hydrophilic bile acids prevent liver damage caused by lack of biliary phospholipid in Mdr2−/− mice. J. Lipid Res. 2019, 60, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Pinon, M.; Carboni, M.; Colavito, D.; Cisarò, F.; Peruzzi, L.; Pizzol, A.; Calosso, G.; David, E.; Calvo, P.L. Not only Alagille syndrome. Syndromic paucity of interlobular bile ducts secondary to HNF1β deficiency: A case report and literature review. Ital. J. Pediatr. 2019, 45, 27. [Google Scholar] [CrossRef]
- Cui, G.; Martin, R.C.; Jin, H.; Liu, X.; Pandit, H.; Zhao, H.; Cai, L.; Zhang, P.; Li, W.; Li, Y. Up-regulation of FGF15/19 signaling promotes hepatocellular carcinoma in the background of fatty liver. J. Exp. Clin. Cancer Res. 2018, 37, 136. [Google Scholar] [CrossRef]
- Attia, Y.M.; Tawfiq, R.A.; Ali, A.A.; Elmazar, M.M. The FXR Agonist, Obeticholic Acid, Suppresses HCC Proliferation & Metastasis: Role of IL-6/STAT3 Signalling Pathway. Sci. Rep. 2017, 7, 12502. [Google Scholar] [CrossRef]
- Markham, A.; Keam, S.J. Obeticholic Acid: First Global Approval. Drugs 2016, 76, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Odevixibat: First Approval. Drugs 2021, 81, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissmann, F.; Cameron, T.O.; Sidobre, S.; Manlongat, N.; Kronenberg, M.; Briskin, M.J.; Dustin, M.L.; Littman, D.R. Intravascular Immune Surveillance by CXCR6+ NKT Cells Patrolling Liver Sinusoids. PLoS Biol. 2005, 3, e113. [Google Scholar] [CrossRef] [Green Version]
- Deneau, M.R.; Mack, C.; Mogul, D.; Perito, E.R.; Valentino, P.L.; Amir, A.Z.; DiGuglielmo, M.; Draijer, L.G.; El-Matary, W.; Furuya, K.N.; et al. Oral Vancomycin, Ursodeoxycholic Acid, or No Therapy for Pediatric Primary Sclerosing Cholangitis: A Matched Analysis. Hepatology 2021, 73, 1061–1073. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Year | Gene | Protein | Phenotypes | Hepatobiliary Cancers |
|---|---|---|---|---|
| 1998 | ATP8B1 | ATP8B1 | ICP BRIC PFIC 1 | Not reported |
| 1998 | ABCB11 | BSEP | ICP DIC LPAC BRIC PFIC 2 | HCC CCA |
| 1996 | ABCB4 | MDR3 | ICP DIC LPAC PFIC 3 | HCC CCA Gallbladder cancer |
| 2014 | TJP2 | TJP2 | ICP PFIC 4 | HCC CCA |
| 2016 | NR1H4 | FXR | ICP PFIC 5 | Not reported |
| 2017 | MYO5B | MYO5B | BRIC MVID MYO5B-PFIC | Not reported |
| 2019 | USP53 | USP53 protein | BRIC PFIC 7 | Not reported |
| 2019 | KIF12 | KIF12 | PFIC 8 | Not reported |
| 2020 | SLC51A | OSTα-OSTβ | PFIC 6 | Not reported |
| Patient †/Gender/Origins | Age, PFIC Onset | Age, HBC Type and Liver Histology | Gene | Nucleotide Changes | Predicted Consequences | PMID |
|---|---|---|---|---|---|---|
| A/M/NEC | Cho from 3 wk | 21 mo (incidental in explant; AFP 199 ng), at LT | ABCB11 | c.1939delA/c.2012-8T>G | p.G648Vfs*6/splice site disruption | 16871584 |
| B/F/NEC | Cho from 2 wk, hospitalized for evaluation aged 12 wk | 28 mo, at open biopsy; AFP not determined | ABCB11 | c.2178 + 1G>A/c.74C>A | Splice site disruption/p.S25* | 16871584 |
| C/M/NEC | Cho from birth | 23 mo (AFP 30k ng; liver mass); histologic diagnosis at necropsy, 24 mo | ABCB11 | c.1445A>G/c.3691C>T | p.D482G/p.R1231W | 16871584 |
| D/M/NEC | Cho from 3 wk | 22 mo (AFP 158k ng); liver mass; lung and bone lesions; chemotherapy given; histologic diagnosis at LT, 25 mo | ABCB11 | c.890A>G/c.890A>G | p.E297G/p.E297G | 16871584 |
| E/M/NEC | Growth failure from 6 mo; diagnosed 9.5 mo | 29 mo (incidental in explant; AFP 6.4k ng), at LT | ABCB11 | c.611 + 1G>A/c.890A>G | Splice site disruption/p.E297G | 16871584 |
| F/M/NEC | Cho from 6 wk | 16 mo (clinically unsuspected), at necropsy; AFP not determined | ABCB11 | c.908 + 1G>A/not known | Splice site disruption/not known | 16871584 |
| G/F/A | Cho from 6 wk | 15 mo, HCC (AFP 11k ng); histologic diagnosis at LT, 16 mo | ABCB11 | c.1416T>A/c.1416T>A | p.Y472*/p.Y472* | 16871584 |
| H/M/NEC | Evaluation at 6 mo for J and growth failure | 52 mo (marked increase in abdominal size; tumour metastasized at diagnosis; AFP 2 × 106 ng), at open biopsy | ABCB11 | c.890A>G/IVS13del-13ˆ-8 | p.E297G/splice site disruption | 16871584 |
| I/M/CAC | Cho from birth | 13 mo (incidental in explant; AFP 831 ng), at LT | ABCB11 | c.2343 + 2T>C/c.2343 + 2T>C | Splice site disruption | 16871584 |
| J/M/CAC | Cho from 1 wk | 14 mo (AFP 4k ng; liver mass), at biopsy; confirmed at LT, 15 mo | ABCB11 | c.2316T>G/c.2316T>G | p.Y772*/p.Y772* | 16871584 |
| K/M/NEC | Cho from 3 mo | 26 mo; HCC metastasized at diagnosis (AFP not reported); at biopsy | ABCB11 | None sought | None predicted | 16871584 |
| A/F/Hi | 2 mo | Giant cell hepatitis and mild portal-tract fibrosis, biliary Cir (3 years), CCA at 4/6/12 yrs | ABCB11 | c.1723C>T/12.5 Mb del | p.R575*/12.5 Mb del | 17452236 |
| B/F/C | J and I in infancy | Giant cell hepatitis (2 mo), hepatic resection revealed advanced biliary Cir with left-lobe peripheral CCA | ABCB11 | c.890A>G/c.2343 + 1G>T | p.Q297G/splice site disruption | 17452236 |
| 1/-/CA-A * | ABCB11 | c.379delA/c.379delA | p.T127Hfs*6/p.T127Hfs*6 | 18395098 | ||
| 7/-/CA-A * | ABCB11 | c.3213 + 1delG/c.3213 + 1delG | Splice defect | 18395098 | ||
| 65 | ABCB11 | c.3382C>T/c.3382C>T | p.R1128C/p.R1128C | 18395098 | ||
| 45a/-/EU | ABCB11 | c.1238T>G/c.1238T>G | p.L413W/p.L413W | 18395098 | ||
| 45b/-/EU | ABCB11 | c.1238T>G/c.1238T>G | p.L413W/p.L413W | 18395098 | ||
| 47a/-/EU | ABCB11 | c.149T>C/c.149T>C | p.L50S/p.L50S | 18395098 | ||
| 47b/-/EU | ABCB11 | c.149T>C/c.149T>C | p.L50S/p.L50S | 18395098 | ||
| 83/-/EU | ABCB11 | c.937C>A/c.1445A>G | p.R313S/p.D482G | 18395098 | ||
| 105/-/EU | ABCB11 | c.1445A>G/not identified | p.D482G/not identified | 18395098 | ||
| 5a/-/- | 10 mo | 4 yrs; H, S, I, J, LF; trifocal HCC, AFP 931 ng | ABCB11 | Not reported | p.Y354*/p.G982R | 20232290 |
| 16/-/- | 1 mo | H, I, recurrent J, LF, AFP 1500 ng (17 mo) | ABCB11 | Not reported | p.R1231W/p.I528* | 20232290 |
| 24/-/- | 1 mo | H, S, permanent J, I, bifocal HCC, normal values of AFP (10 yrs), LF (12 yrs) | ABCB11 | Not reported | p.R1153C/c.3213 + 4A>G | 20232290 |
| 27/-/- | 3 wk | H, I, decreased J, AFP 5000 ng (10 yrs), HCC (1 nodule resected) | ABCB11 | Not reported | p.G982R/p.R1001R (predicted to affect splicing) | 20232290 |
| 32/-/- | 1 mo | H, pemanent J, I, HCC (2 nodules), AFP 3600 ng, LF (7 mo) | ABCB11 | Not reported | p.R698H/nf | 20232290 |
| 33/-/- | 1 wk | H, permanent J, DS, I AFP 124752 (2mo) and 19770 ng (5mo), no nodule, LF (4 mo) | ABCB11 | Not reported | p.R698H/nf | 20232290 |
| 1/F/- | P at the age of 8 mo | 8 mo | ABCB11 | Not reported | p.A389P/p.R1226H | 25016225 |
| 10/F/EU | 54 yrs | Periportal fibrosis, mild ductular reaction, steatosis, biliary dysplasia, CCA | ABCB4 | c.1005 + 5G>A/wt | Splicing/wt | 22331132 |
| 13/F/EU | 55 yrs | Biliary Cir, macronodule with well-differentiated HCC | ABCB4 | c.959C>T/wt | p.S320F/wt | 22331132 |
| GWAS Icelandic population | <40 yrs | Liver, gallbladder and gallways cancer OR 2.42 | ABCB4 | c.1865G>A | p.G622E | 25807286 |
| GWAS Icelandic population | <40 yrs | Liver, gallbladder and gallways cancer OR 3.07 | ABCB4 | c.1333_1334delCT | p.L445Gfs*22 | 25807286 |
| GWAS Icelandic population | <40 yrs | Liver, gallbladder and gallways cancer OR 4.75 | ABCB4 | c.1529A>G | p.N510S | 25807286 |
| GWAS Icelandic population | <40 yrs | Liver, gallbladder and gallways cancer OR 0.99 | ABCB4 | c.711A>T | p.I237= | 25807286 |
| 1/-/EU | LPAC | CCA diagnosed | ABCB4 | c.1405A>T/wt | p.R469W/wt | 32893960 |
| 2/-/EU | LPAC | CCA diagnosed | ABCB4 | c.1268A>C/wt | p.Q423P/wt | 32893960 |
| 3/-/EU | LPAC | HCC diagnosed | ABCB4 | c.760G>A/wt; c.1546A>G/wt; c.2363G>A/wt | p.A254T/wt; p.M516V/wt; p.R788Q/wt | 32893960 |
| 1/F/C | Neonatal onset, 26 mo | At liver biopsy moderately differentiated HCC in a chronic Cho with Cir | TJP2 | c.2668-1G>T/c.2438dupT | Splice defect/p.N814Qfs*28 | 25921221 |
| 2/M/C | Neonatal onset, 6 mo | Liver biopsy at 6 mo showed C, giant cell transformation, and micronodular Cir | TJP2 | c.817delG/c.817delG | p.A273Pfs*38/p.A273Pfs*38 | 25921221 |
| 1/F/- | 1 month | 7 yrs | TJP2 NM_001170416.1 | c.(2659 + 1_2660-1)_(2760 + 1_2761-1)del/c.(2659 + 1_2660-1)_(2760 + 1_2761-1)del | Skipping exon 18/skipping exon 18 | 28733884 |
| P2.5/M/- | 20 yrs | 23 yrs | TJP2 | c.3334C>T/c.3334C>T | p.Q1112*/p.Q1112* | 32089630 |
| 1/M/- | ALGS at 13 wk of age | 3 yrs | JAG1 | De novo mutation of the protein-encoding JAG1-region of chr. 20 (not shown by the authors) | 20714715 | |
| -/F/- | ALGS around age 35 yrs | JAG1 | c.693_694del state not specified | p.Arg231Serfs*10 | 33369123 | |
| 1/-/- | Cir, 1.5 y | JAG1 | c.551G>A/wt | p.R184H/wt | 32180165 | |
| 2/-/- | Cir, 2 yrs | NOTCH2 | c.5830G>A/c.5830G>A | p.G1944S/p.G1944S | 32180165 | |
| 1/F/Ja | CTLDN2; 40 yrs, after her first baby | 40 yrs | SLC25A13 | c.1180 + 1G>A/c.1180 + 1G>A | Splice site disruption | 14606711 |
| 1/M/- | No symptoms | 50 yrs, HCC and intrahepatic CCA | SLC25A13 | c.1180 + 1G>A/c.1180 + 1G>A | Splice site disruption | 18385606 |
| 1/M/T | CTLDN2; 34 yrs | HCC at 48 yrs | SLC25A13 | c.852_855del/c.852_855del | p.M285Pfs*2/p.M285Pfs*2 | 17000460 |
| 1/M/Ch | After birth | HCC at 6 yrs | SLC25A13 | c.852_855del/c.852_855del | p.M285Pfs*2/p.M285Pfs*2 | 30591617 |
| 1/M/- | After birth | HCC at 16 mo | HNF1B | 1.5 Mb deletion of chr. 17/wt | No protein/wt | 29727438 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, G.; Mattiaccio, A.; Conti, A.; Turco, L.; Seri, M.; Piscaglia, F.; Morelli, M.C. Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature. Cancers 2022, 14, 3421. https://doi.org/10.3390/cancers14143421
Vitale G, Mattiaccio A, Conti A, Turco L, Seri M, Piscaglia F, Morelli MC. Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature. Cancers. 2022; 14(14):3421. https://doi.org/10.3390/cancers14143421
Chicago/Turabian StyleVitale, Giovanni, Alessandro Mattiaccio, Amalia Conti, Laura Turco, Marco Seri, Fabio Piscaglia, and Maria Cristina Morelli. 2022. "Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature" Cancers 14, no. 14: 3421. https://doi.org/10.3390/cancers14143421