
Targeting Immunosuppressive Tumor-Associated Macrophages Using Innate T Cells for Enhanced Antitumor Reactivity

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies and Flow Cytometry
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Generation of Human M2-Polarized Macrophage
2.5. Generation of Human Mucosal-Associated Invariant T (MAIT), Invariant Natural Killer T (iNKT), Gamma Delta T (γδT), and Conventional Alpha Beta T (αβT) Cells
2.6. Generation of Human Mesothelin CAR-Engineered αβT (MCAR-αβT) and Mesothelin CAR-Engineered MAIT (MCAR-MAIT) cells
2.7. In Vitro Tumor Cell Killing Assay
2.8. In Vitro Mixed Lymphocyte Reaction (MLR) Assay
2.9. In Vitro Mixed Mφ/T Reaction Assay
2.10. 3D Tumor/TAM/T-Cell Organoid Culture
2.11. Statistical Analysis
3. Results
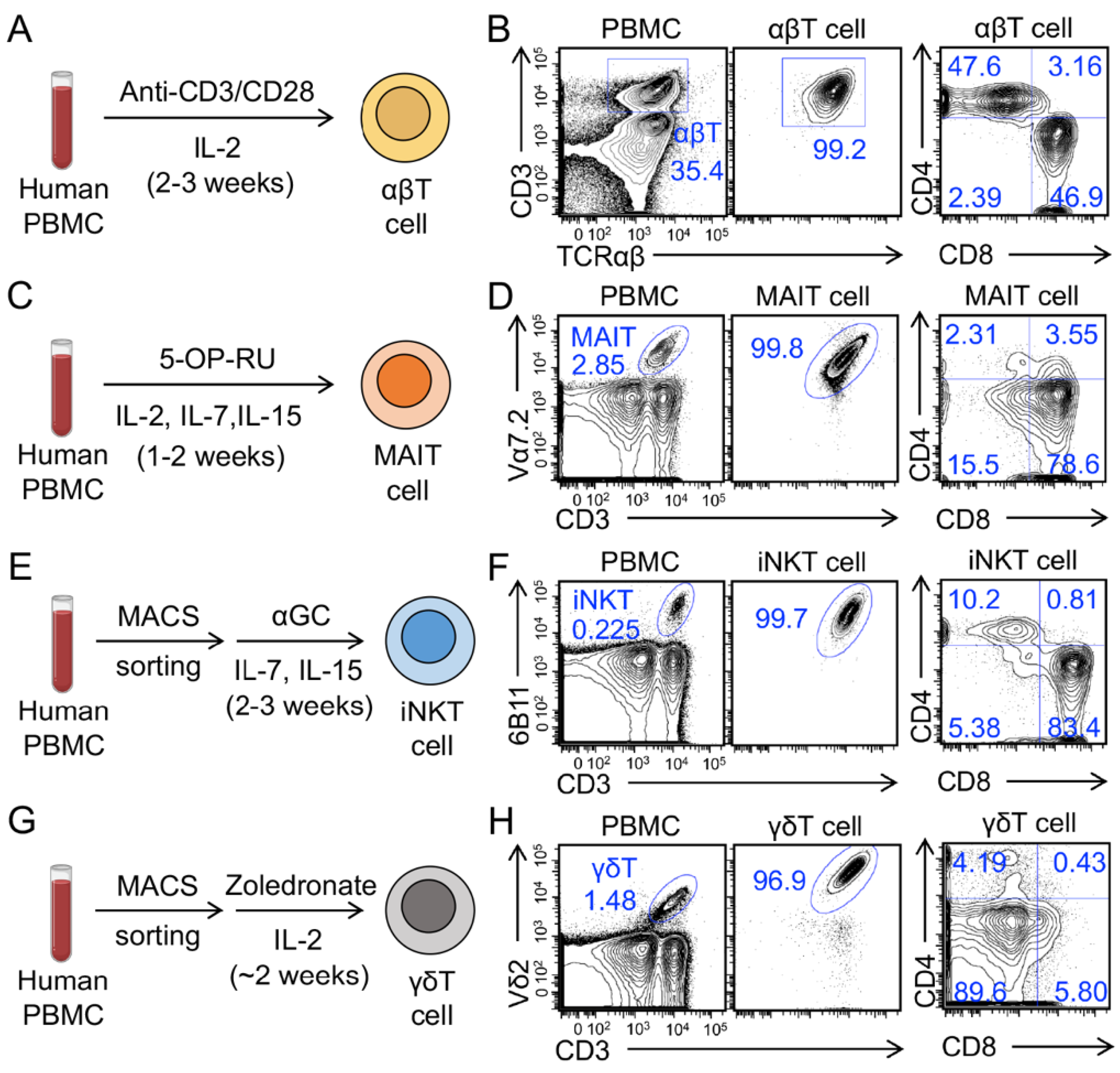
3.1. Generation and Characterization of Human Peripheral Blood Mononuclear Cell (PBMC)-Derived Innate T Cells
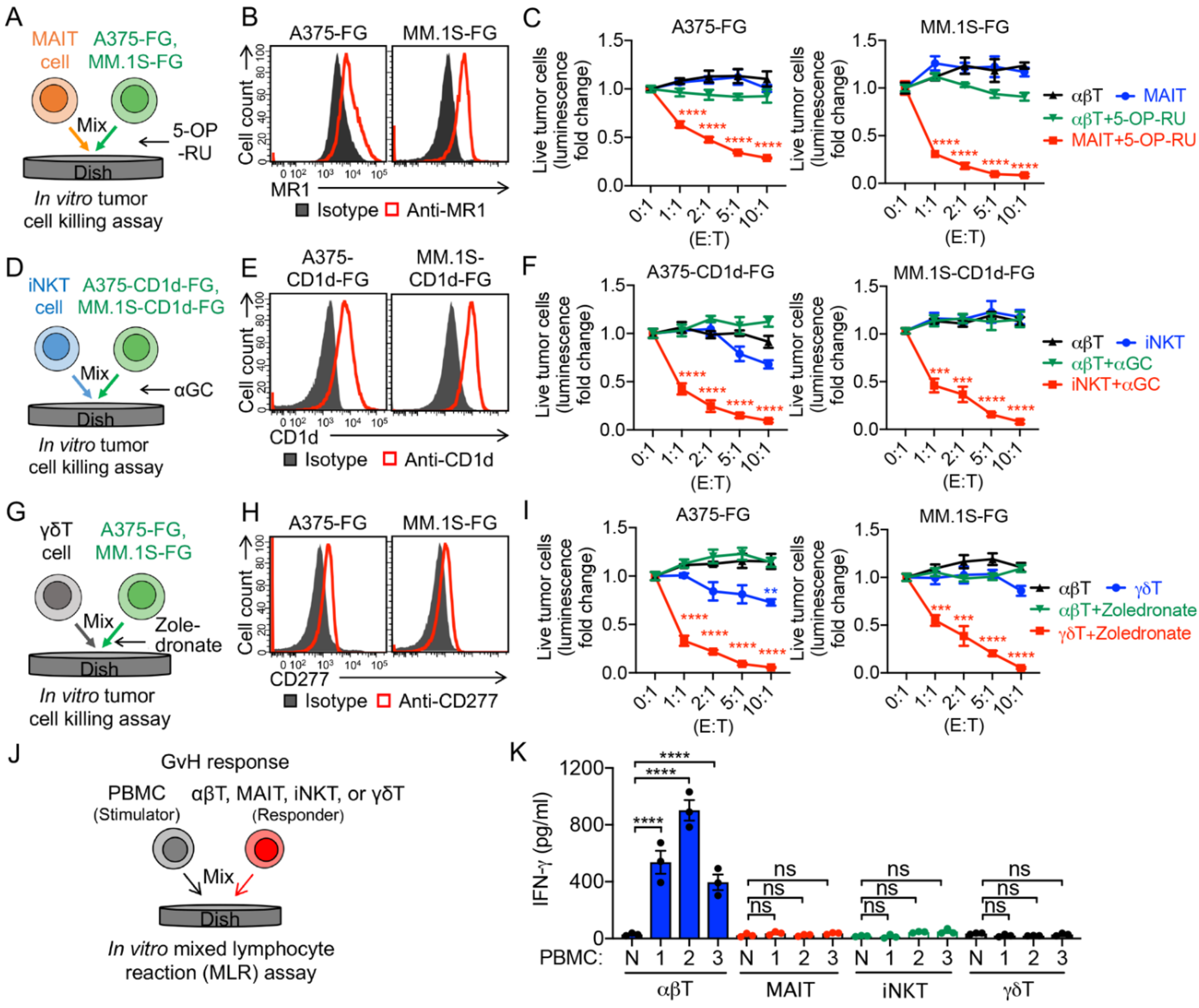
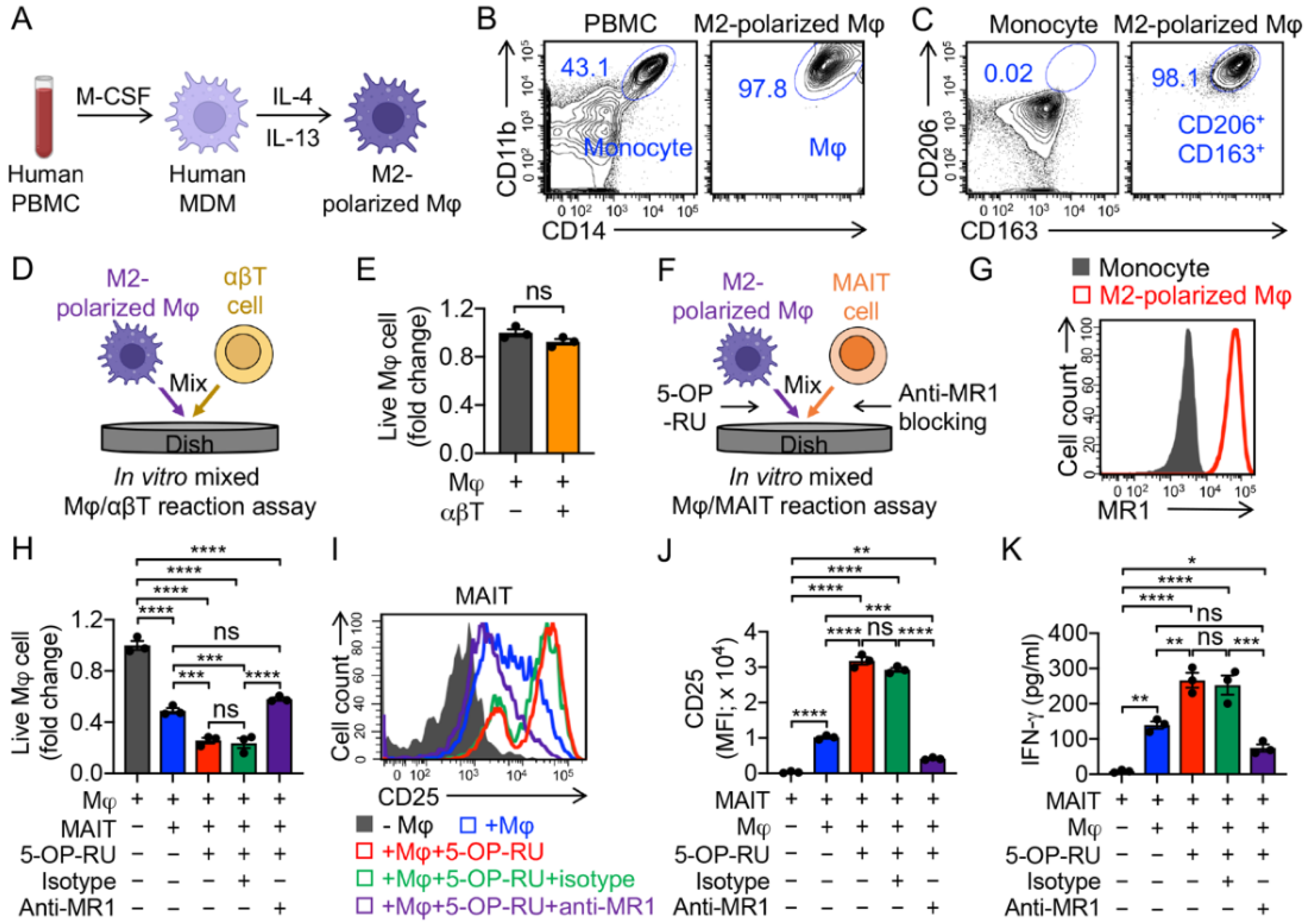
3.2. Targeting Immunosuppressive M2-Polarized Macrophages by MAIT Cells
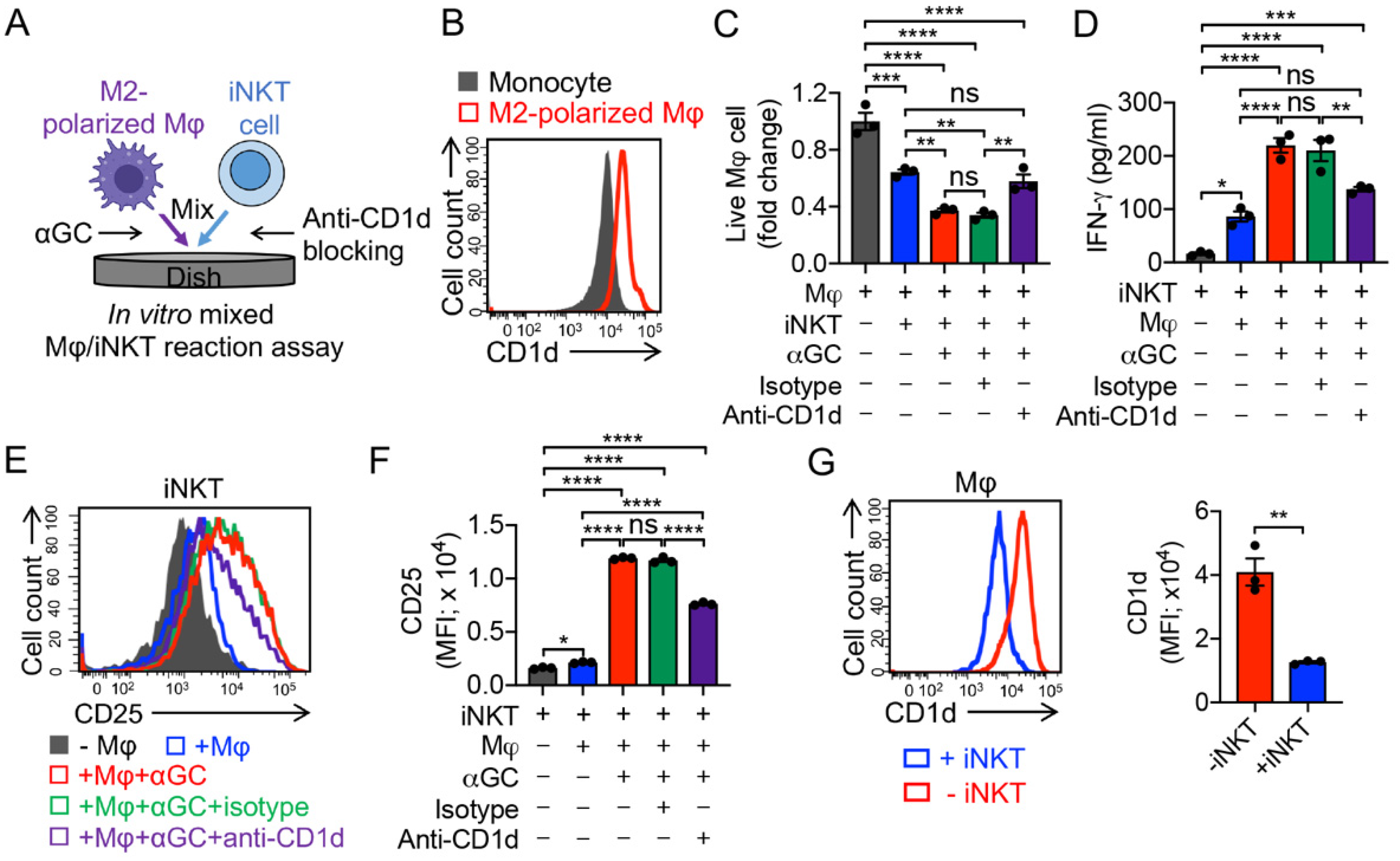
3.3. Targeting Immunosuppressive M2-Polarized Macrophages by iNKT Cells
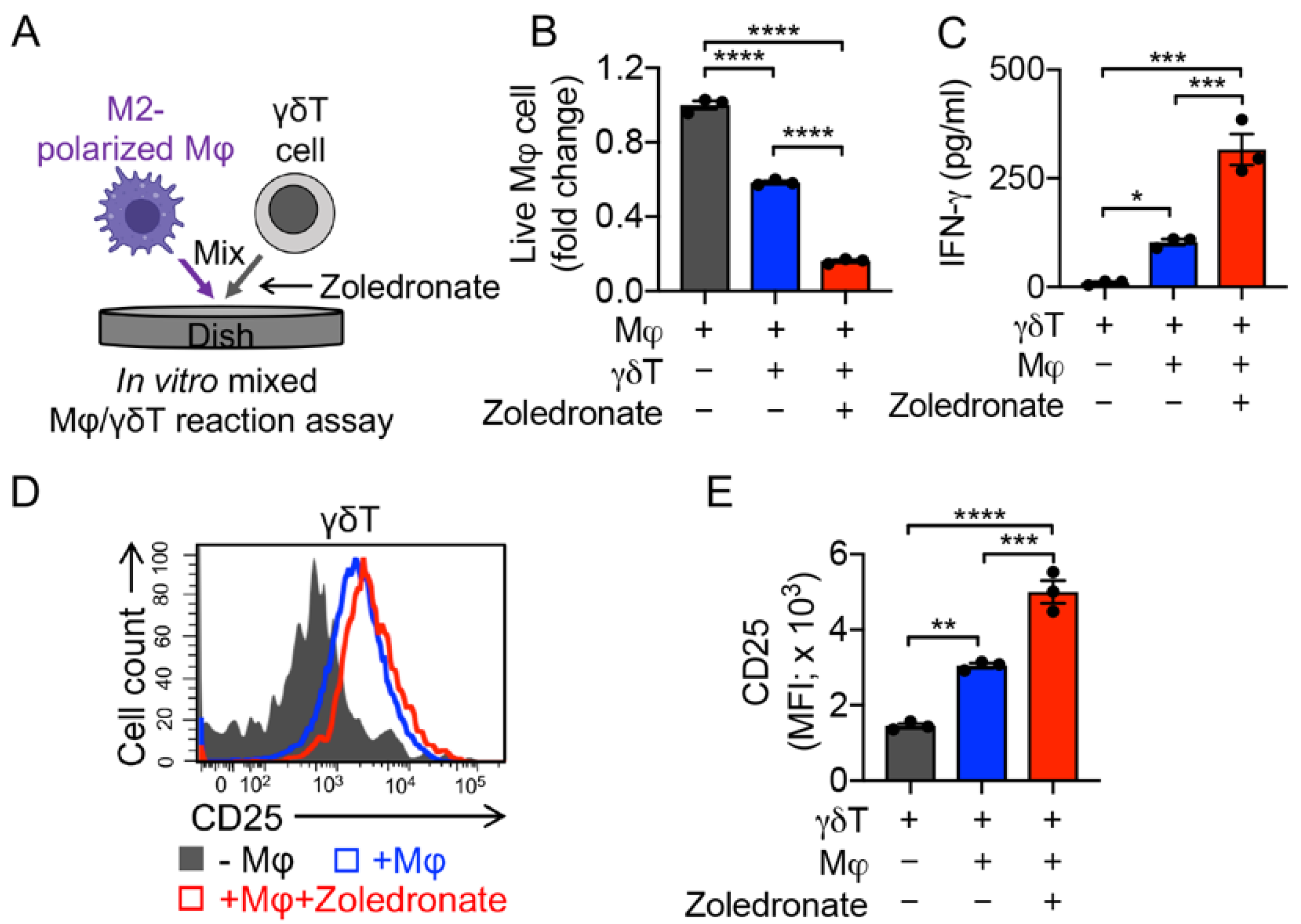
3.4. Targeting Immunosuppressive M2-Polarized Macrophages by γδT Cells
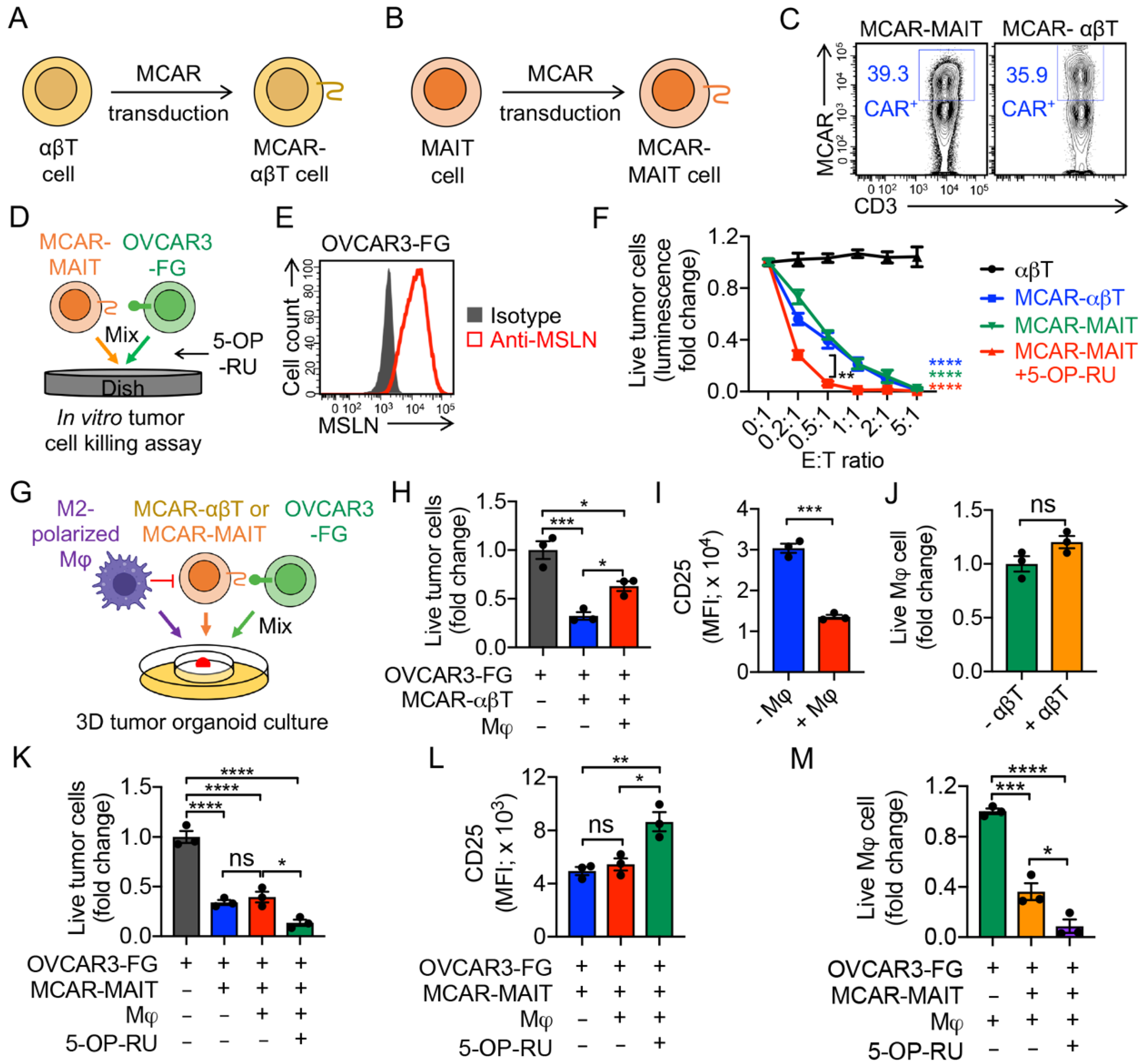
3.5. Targeting Tumor-Associated Macrophages (TAMs) by Mesothelin-Targeting CAR (MCAR)-Engineered MAIT (MCAR-MAIT) Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Gao, Y.; Williams, A.P. Role of Innate T Cells in Anti-Bacterial Immunity. Front. Immunol. 2015, 6, 302. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-R.; Dunn, Z.S.; Zhou, Y.; Lee, D.; Yang, L. Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells 2021, 10, 3497. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kramer, A.; Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 2021, 7, 1059–1073. [Google Scholar] [CrossRef]
- Lamichhane, R.; Ussher, J.E. Expression and trafficking of MR1. Immunology 2017, 151, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Motozono, C.; Hattori, A.; Kakeya, H.; Yamasaki, S.; Oishi, S.; Ohno, H.; Inuki, S. The effects of 5-OP-RU stereochemistry on its stability and MAIT-MR1 axis. Chembiochem 2021, 22, 672–678. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant natural killer T cells: Bridging innate and adaptive immunity. Cell Tissue Res. 2011, 343, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, G.; Zhang, J.; Wu, X.; Chen, X. The Dual Roles of Human γδ T Cells: Anti-Tumor or Tumor-Promoting. Front. Immunol. 2020, 11, 619954. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, S.; Elahi, R.; Esmaeilzadeh, A. Solid Tumors Challenges and New Insights of CAR T Cell Engineering. Stem Cell Rev. Rep. 2019, 15, 619–636. [Google Scholar] [CrossRef]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2021, 1499, 18–41. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Li, Y.-R.; Yu, Y.; Kramer, A.; Hon, R.; Wilson, M.; Brown, J.; Yang, L. An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells 2022, 11, 1583. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Wang, X.; Yu, J.; Ma, F.; Li, Z.; Zhou, Y.; Zeng, S.; Ma, X.; Li, Y.-R.; Neal, A.; et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat. Commun. 2021, 12, 3530. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Li, B.; Yang, L. MAOI Antidepressants: Could They Be a Next-Generation ICB Therapy? Front. Immunol. 2022, 13, 853624. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Dunn, Z.S.; Garcia, G., Jr.; Carmona, C.; Zhou, Y.; Lee, D.; Yu, J.; Huang, J.; Kim, J.T.; Arumugaswami, V.; et al. Development of off-the-shelf hematopoietic stem cell-engineered invariant natural killer T cells for COVID-19 therapeutic intervention. Stem Cell Res. Ther. 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Hanafi, L.A.; Sheih, A.; Golob, J.L.; Srinivasan, S.; Boeckh, M.J.; Pergam, S.A.; Mahmood, S.; Baker, K.K.; Gooley, T.A.; et al. Graft-Derived Reconstitution of Mucosal-Associated Invariant T Cells after Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Gherardin, N.A.; Loh, L.; Admojo, L.; Davenport, A.J.; Richardson, K.; Rogers, A.; Darcy, P.K.; Jenkins, M.R.; Prince, H.M.; Harrison, S.J.; et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Garner, L.C.; Klenerman, P.; Provine, N.M. Insights into Mucosal-Associated Invariant T Cell Biology from Studies of Invariant Natural Killer T Cells. Front. Immunol. 2018, 9, 1478. [Google Scholar] [CrossRef]
- Kurioka, A.; Jahun, A.S.; Hannaway, R.F.; Walker, L.J.; Fergusson, J.R.; Sverremark-Ekström, E.; Corbett, A.J.; Ussher, J.E.; Willberg, C.B.; Klenerman, P. Shared and distinct phenotypes and functions of human cD161++ Vα7.2+ T cell subsets. Front. Immunol. 2017, 8, 1031. [Google Scholar] [CrossRef]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [Green Version]
- Garcillán, B.; Marin, A.V.M.; Jiménez-Reinoso, A.; Briones, A.C.; Muñoz-Ruiz, M.; García-León, M.J.; Gil, J.; Allende, L.M.; Martínez-Naves, E.; Toribio, M.L.; et al. γδ T Lymphocytes in the Diagnosis of Human T Cell Receptor Immunodeficiencies. Front. Immunol. 2015, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The role of natural killer T cells in cancer-A phenotypical and functional approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Oh, S.; Lim, S.; Shin, J.H.; Yoon, M.S.; Park, S.-H. Invariant NKT cells facilitate cytotoxic T-cell activation via direct recognition of CD1d on T cells. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.A.; Seo, H.; Kim, I.K.; Jeon, I.; Kang, C.Y. Roles of NKT cells in cancer immunotherapy. Arch. Pharm. Res. 2019, 42, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Takahashi, T.; Hiruma, K.; Kanda, Y.; Tanaka, Y.; Ogawa, S.; Chiba, S.; Miura, O.; Sakamaki, H.; Hirai, H. Recovery of Valpha24+ NKT cells after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004, 34, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Bohineust, A.; Tourret, M.; Derivry, L.; Caillat-Zucman, S. Mucosal-associated invariant T (MAIT) cells, a new source of universal immune cells for chimeric antigen receptor (CAR)-cell therapy. Bull. Cancer 2021, 108, S92–S95. [Google Scholar] [CrossRef]
- Morandi, F.; Yazdanifar, M.; Cocco, C.; Bertaina, A.; Airoldi, I. Engineering the Bridge between Innate and Adaptive Immunity for Cancer Immunotherapy: Focus on γδ T and NK Cells. Cells 2020, 9, 1757. [Google Scholar] [CrossRef]
- Makkouk, A.; Yang, X.C.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Jin, X.; Kruth, H.S. Culture of Macrophage Colony-stimulating Factor Differentiated Human Monocyte-derived Macrophages. J. Vis. Exp. 2016, 112, e54244. [Google Scholar] [CrossRef]
- Nielsen, M.C.; Hvidbjerg Gantzel, R.; Clària, J.; Trebicka, J.; Møller, H.J.; Grønbæk, H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells 2020, 9, 1175. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch′ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinks, T.S.C.; Zhang, X.-W. MAIT Cell Activation and Functions. Front. Immunol. 2020, 11, 1014. [Google Scholar] [CrossRef] [PubMed]
- Joana, D.; Caroline, B.; Jean-Baptiste, G.A.; van den Biggelaar, R.H.G.; Lal, K.G.; Anna, G.; Liyen, L.; Yaaseen, G.M.; Rong, S.W.; Sudipto, B.; et al. The CD4−CD8− MAIT cell subpopulation is a functionally distinct subset developmentally related to the main CD8+ MAIT cell pool. Proc. Natl. Acad. Sci. USA 2018, 115, E11513–E11522. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018, 34, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, K.; Arimoto, A.; Nishi, M.; Tanaka, T.; Fujita, M.; Fukuoka, E.; Sugita, Y.; Nakagawa, A.; Hasegawa, H.; Suzuki, S.; et al. Application of iNKT Cell-targeted Active Immunotherapy in Cancer Treatment. Anticancer Res. 2018, 38, 4233–4239. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Sun, R.; Chen, Y.; Wei, H.; Tian, Z. γδT cells drive myeloid-derived suppressor cell-mediated CD8+ T cell exhaustion in hepatitis B virus-induced immunotolerance. J. Immunol. 2014, 193, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [Green Version]
- Qu, P.; Wang, L.-Z.; Lin, P.C. Expansion and functions of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Lett. 2016, 380, 253–256. [Google Scholar] [CrossRef]
- Fowler, D.W.; Copier, J.; Dalgleish, A.G.; Bodman-Smith, M.D. Zoledronic acid causes γδ T cells to target monocytes and down-modulate inflammatory homing. Immunology 2014, 143, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, P.; Shojaei, H.; Schittek, B.; Gieseler, F.; Wollenberg, B.; Kalthoff, H.; Kabelitz, D.; Wesch, D. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: Involvement of NKG2D ligands and T-cell receptor- versus NKG2D-dependent recognition. Scand. J. Immunol. 2007, 66, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Orozco, B.; Kunzmann, V.; Wrobel, P.; Kabelitz, D.; Steinle, A.; Herrmann, T. Activation of V gamma 9V delta 2 T cells by NKG2D. J. Immunol. 2005, 175, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigné, C.-M.; Mönkkönen, H.; Mönkkönen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabelitz, D. CD277 takes the lead in human γδ T-cell activation. Blood 2012, 120, 2159–2161. [Google Scholar] [CrossRef] [Green Version]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- Hoves, S.; Ooi, C.-H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Shirota, H.; Klinman, D.M. Effect of CpG ODN on monocytic myeloid derived suppressor cells. Oncoimmunology 2012, 1, 780–782. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.-P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Chen, J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V. Immunotherapy: Tisagenlecleucel-the first approved CAR-T-cell therapy: Implications for payers and policy makers. Nat. Rev. Clin. Oncol. 2018, 15, 11–12. [Google Scholar] [CrossRef]
- Martin, J.D.; Miyazaki, T.; Cabral, H. Remodeling tumor microenvironment with nanomedicines. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1730. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-R.; Brown, J.; Yu, Y.; Lee, D.; Zhou, K.; Dunn, Z.S.; Hon, R.; Wilson, M.; Kramer, A.; Zhu, Y.; et al. Targeting Immunosuppressive Tumor-Associated Macrophages Using Innate T Cells for Enhanced Antitumor Reactivity. Cancers 2022, 14, 2749. https://doi.org/10.3390/cancers14112749
Li Y-R, Brown J, Yu Y, Lee D, Zhou K, Dunn ZS, Hon R, Wilson M, Kramer A, Zhu Y, et al. Targeting Immunosuppressive Tumor-Associated Macrophages Using Innate T Cells for Enhanced Antitumor Reactivity. Cancers. 2022; 14(11):2749. https://doi.org/10.3390/cancers14112749
Chicago/Turabian StyleLi, Yan-Ruide, James Brown, Yanqi Yu, Derek Lee, Kuangyi Zhou, Zachary Spencer Dunn, Ryan Hon, Matthew Wilson, Adam Kramer, Yichen Zhu, and et al. 2022. "Targeting Immunosuppressive Tumor-Associated Macrophages Using Innate T Cells for Enhanced Antitumor Reactivity" Cancers 14, no. 11: 2749. https://doi.org/10.3390/cancers14112749