
Molecular Characterization of BRCA1 c.5339T>C Missense Mutation in DNA Damage Response of Triple-Negative Breast Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
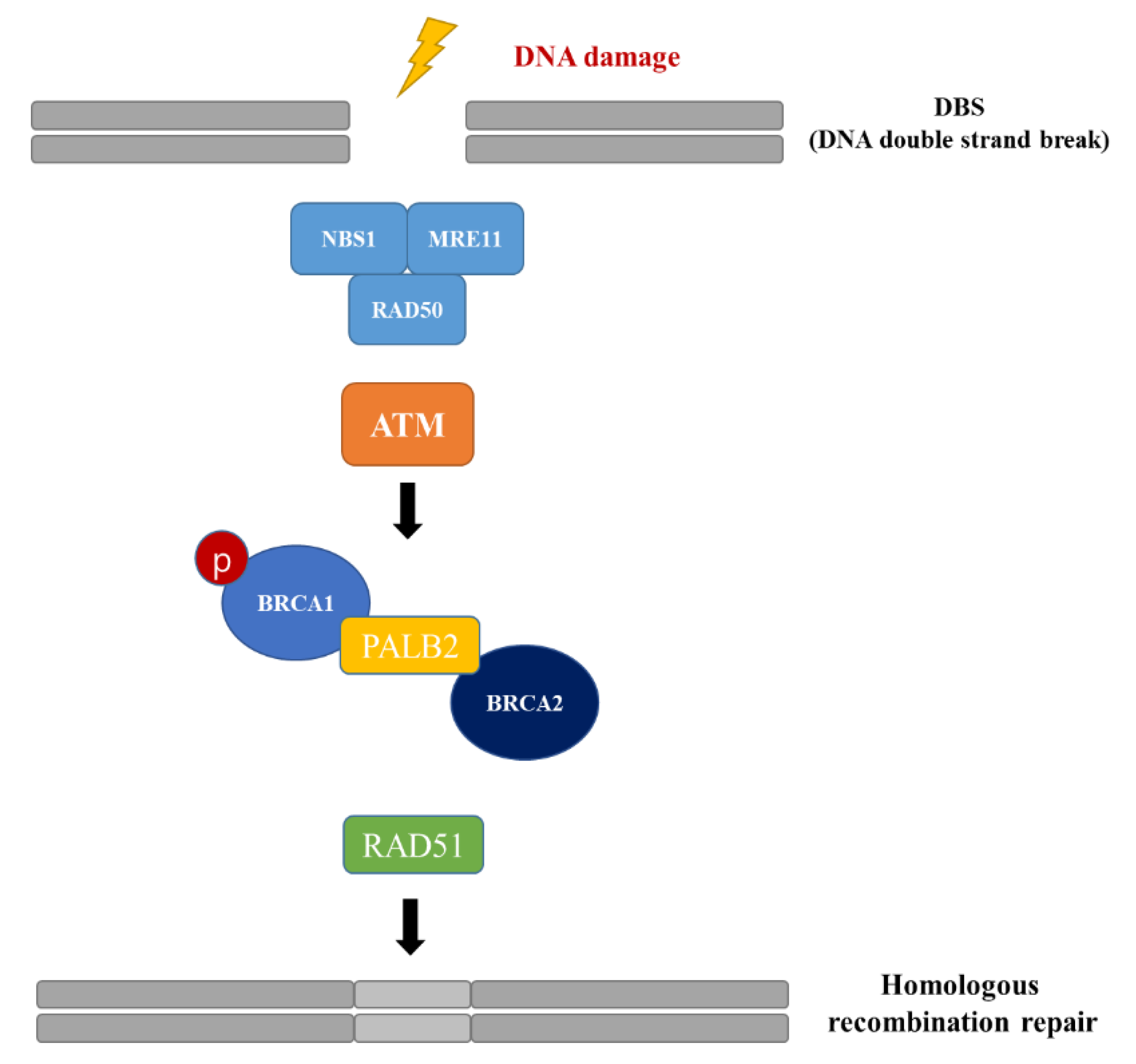
1. Introduction
2. Methods
2.1. Cell Culture
2.2. cDNA Transfection
2.3. Cell Migration Assay (Wound Closure Assay)
2.4. Cell Invasion Assay
2.5. Cell Proliferation Assay
2.6. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.7. Western Blot
2.8. Co-Immunoprecipitation (Co-IP)
2.9. Immunofluorescence (IF)
2.10. Small Interfering RNA (siRNA)
2.11. Olparib Resistant Cell Lines
2.12. Statistical Analysis
3. Results
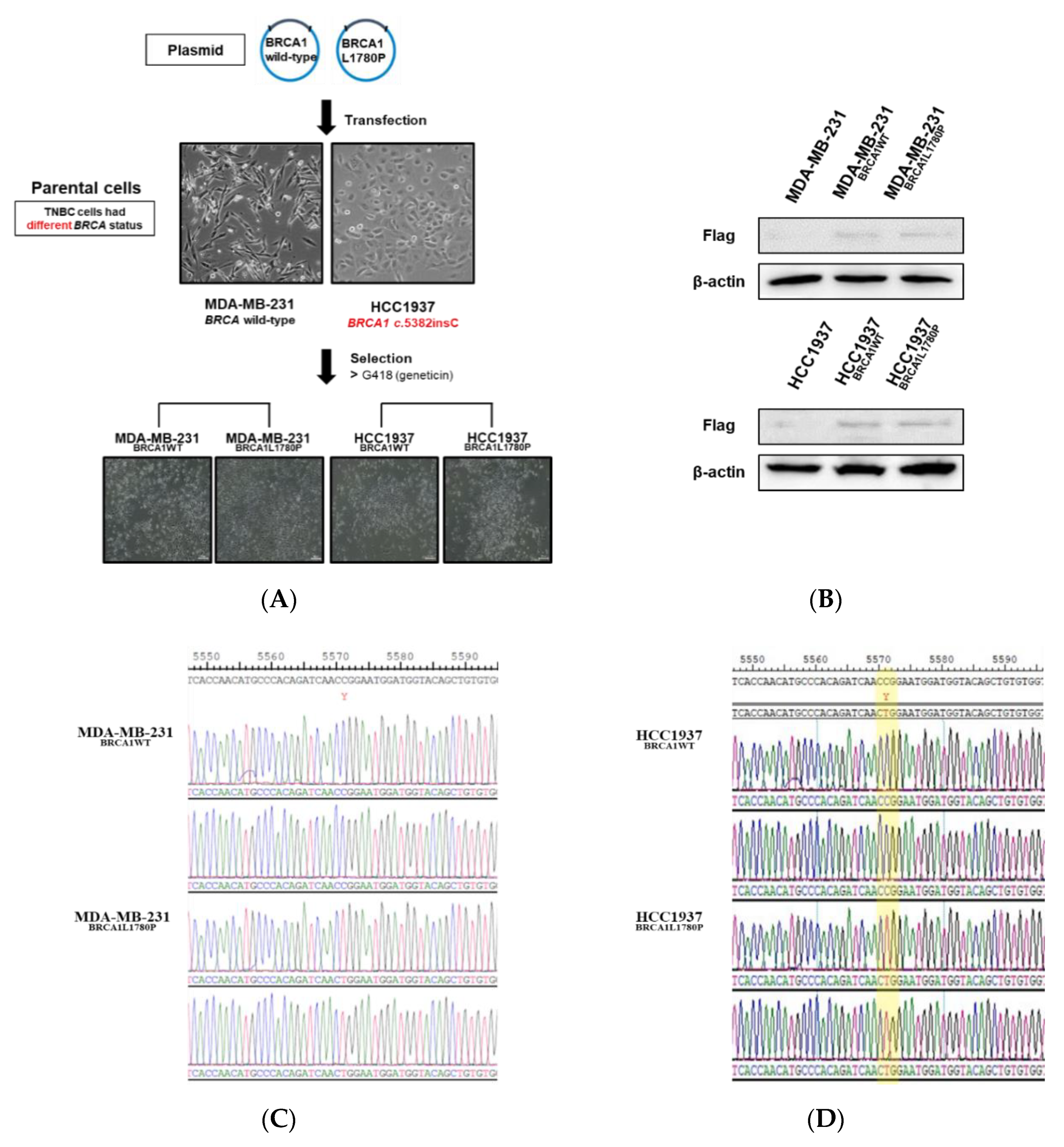
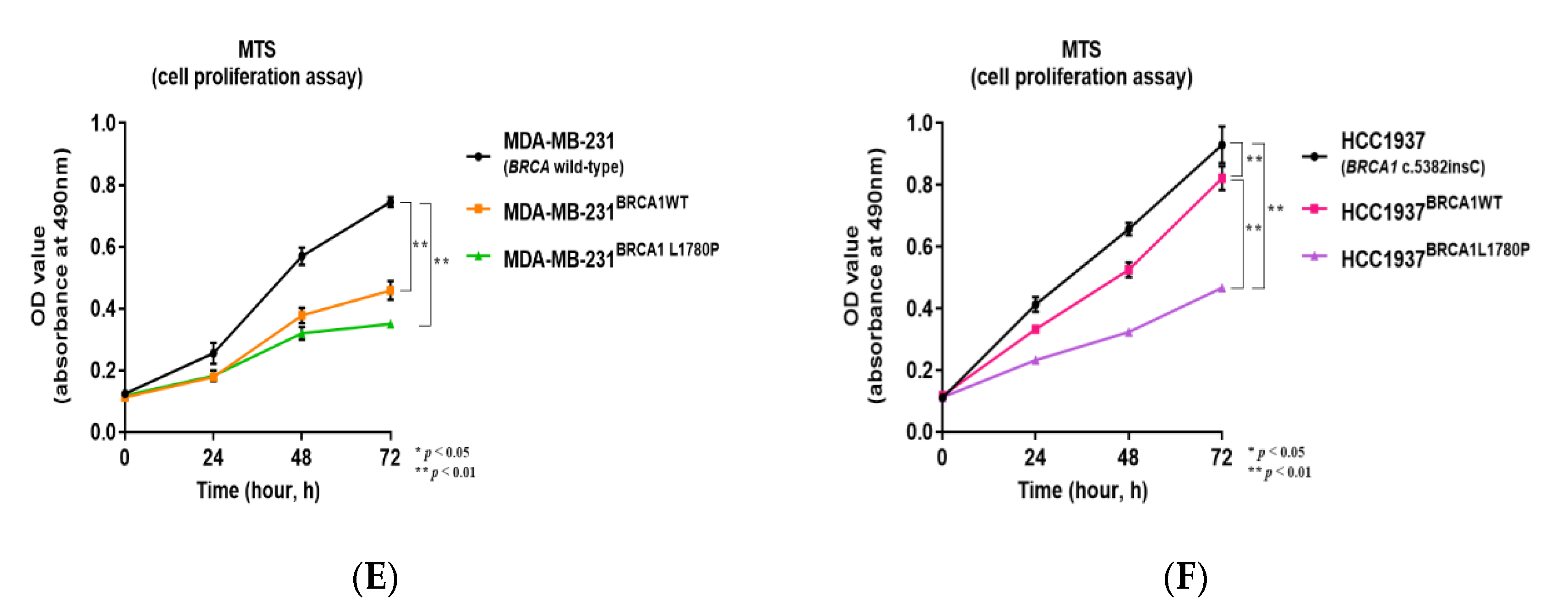
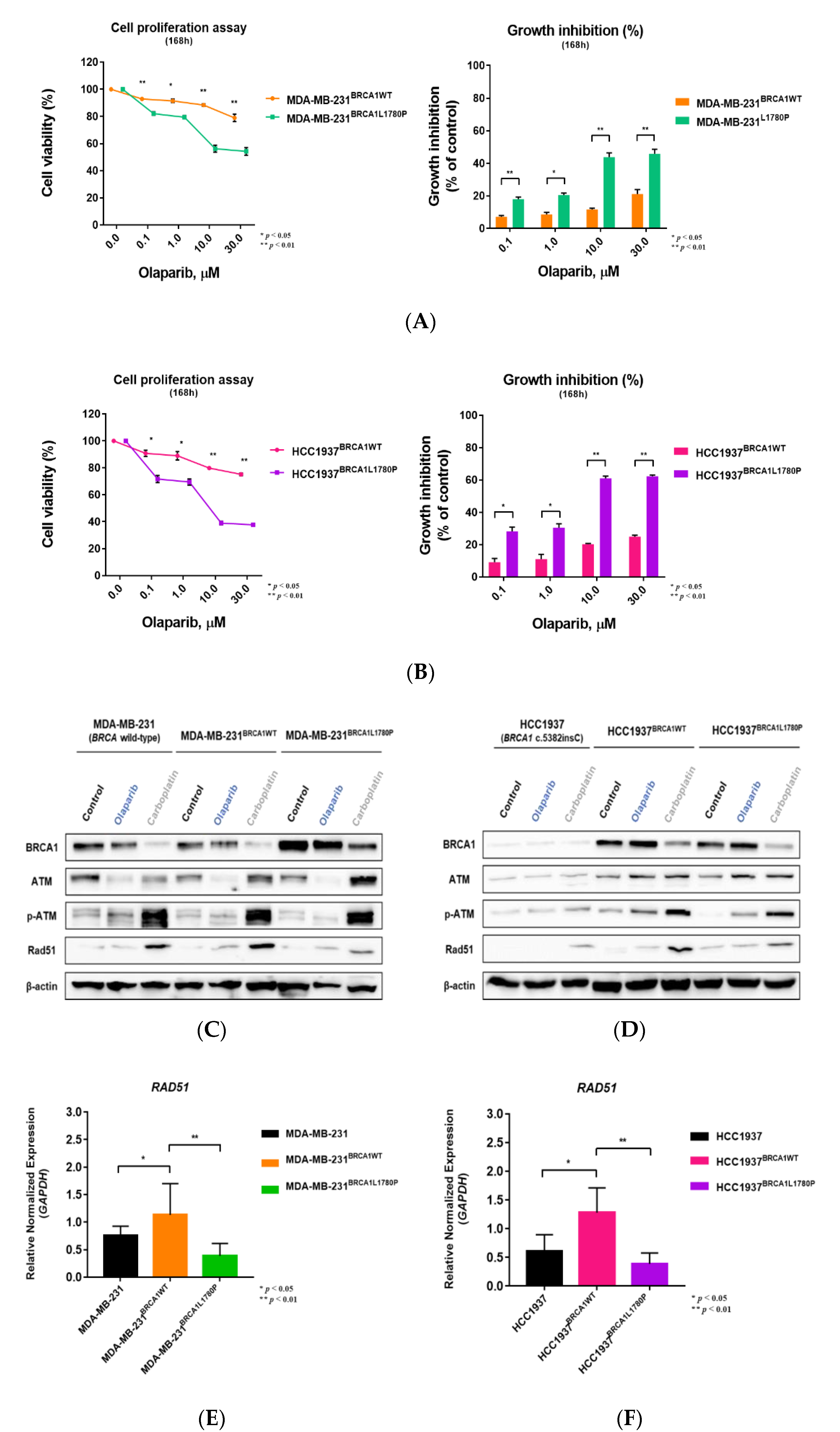
3.1. Stable Expression of Mutant BRCA1 Proteins in TNBC Cells and Its Impact on Cell Proliferation
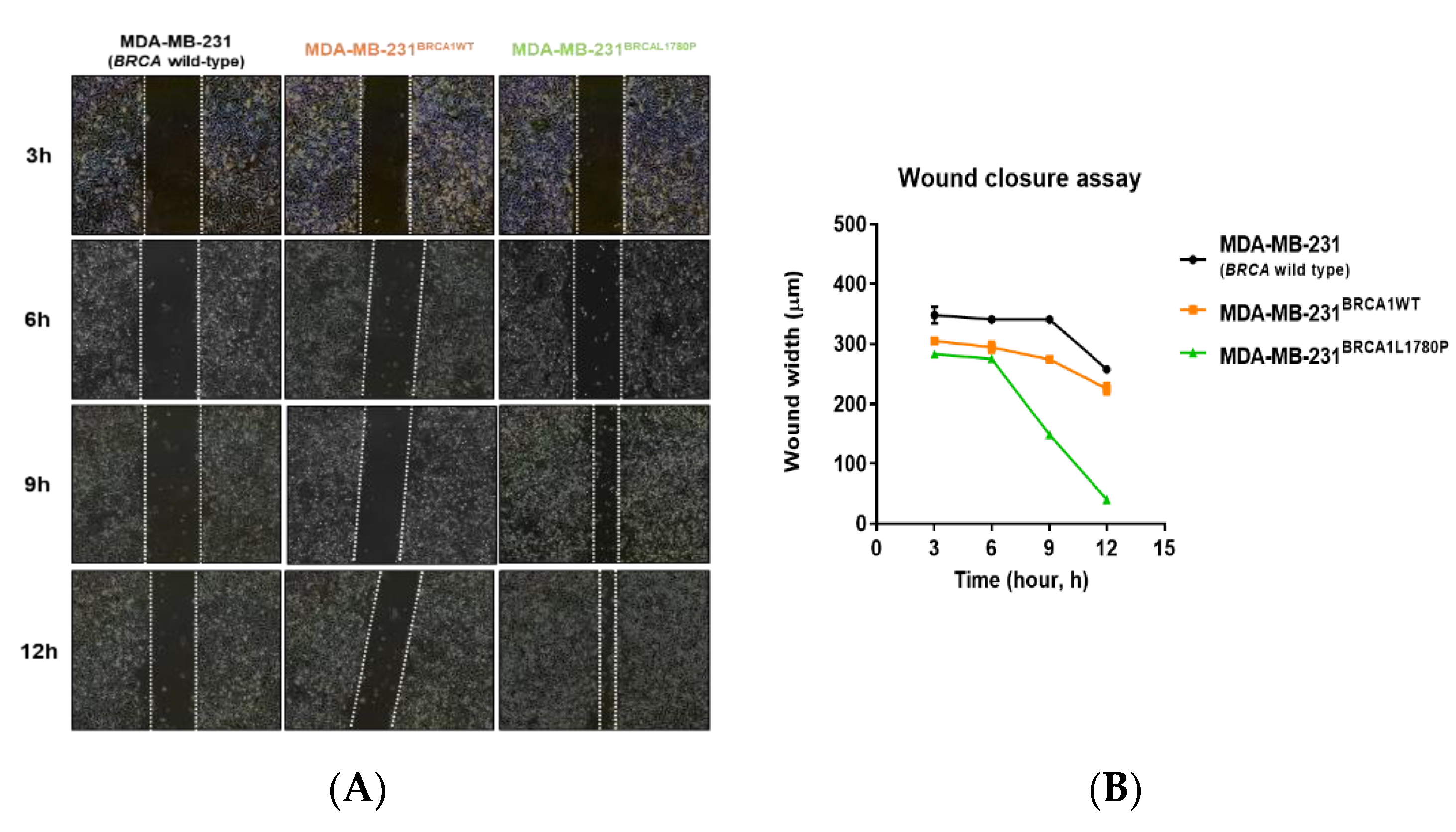
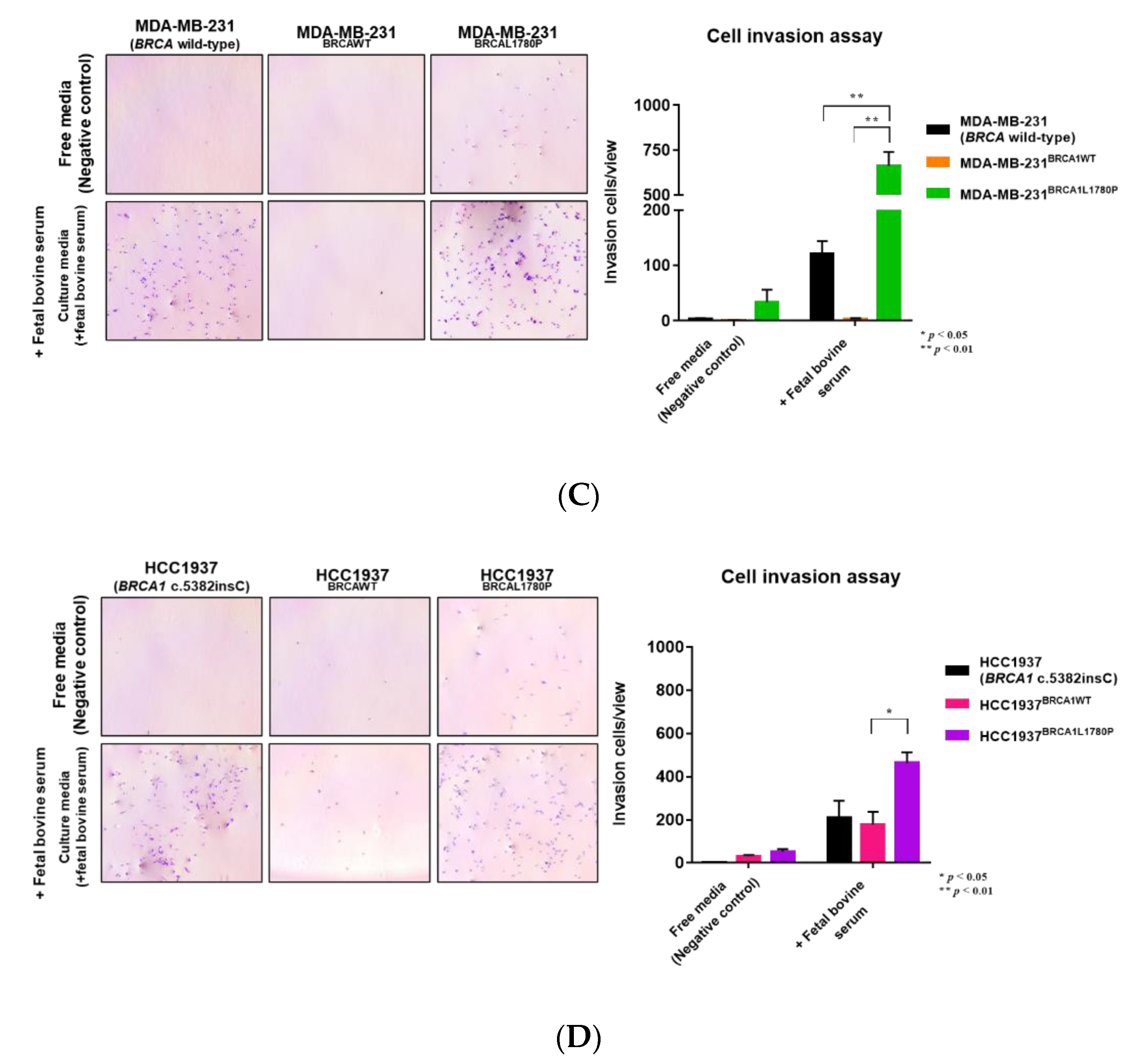
3.2. Cell Migration and Invasion Capacity of BRCA1 L1780P TNBC Cells
3.3. Differential Effect of BRCA1 Mutation Status on Olaparib Sensitivity
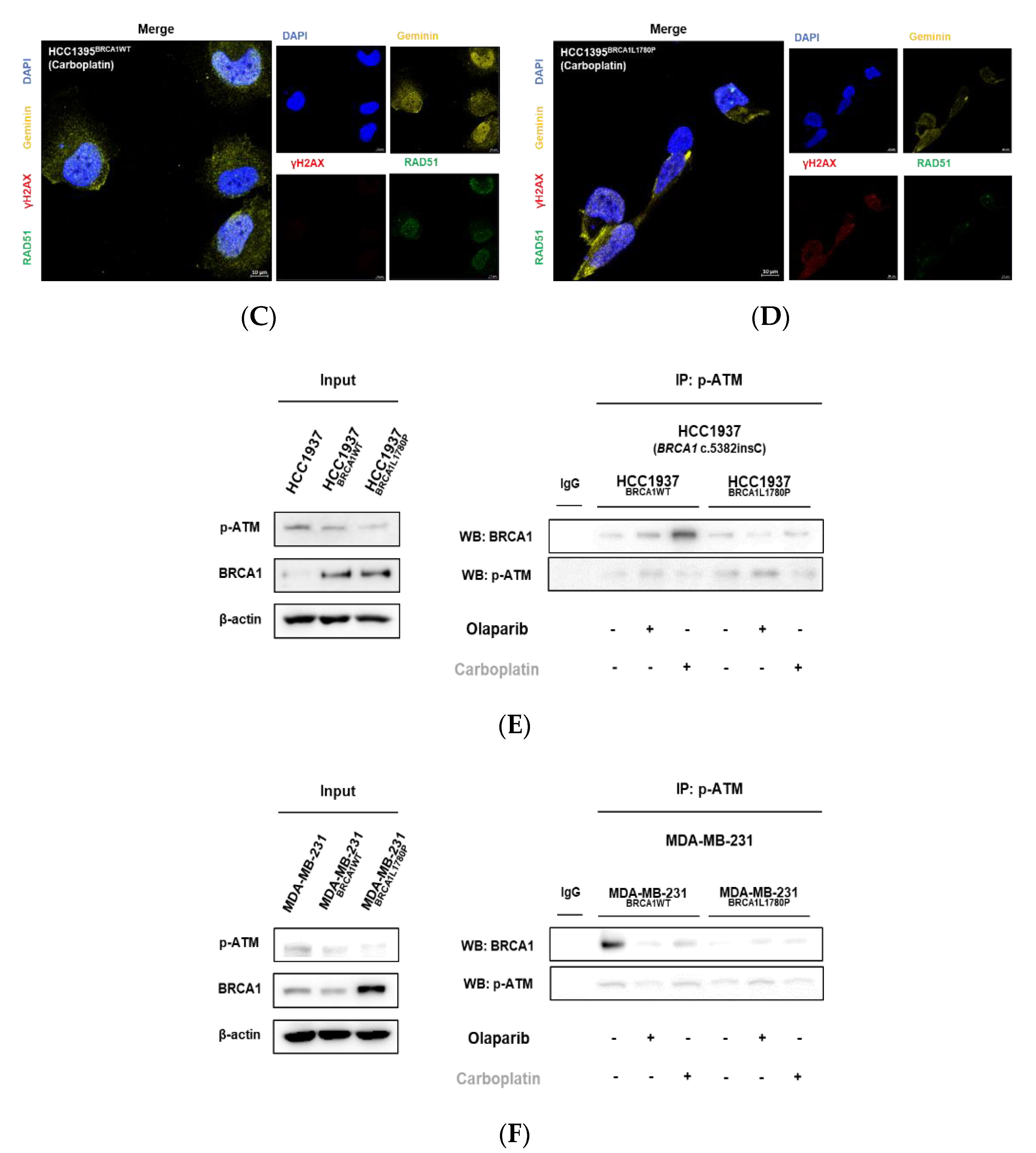
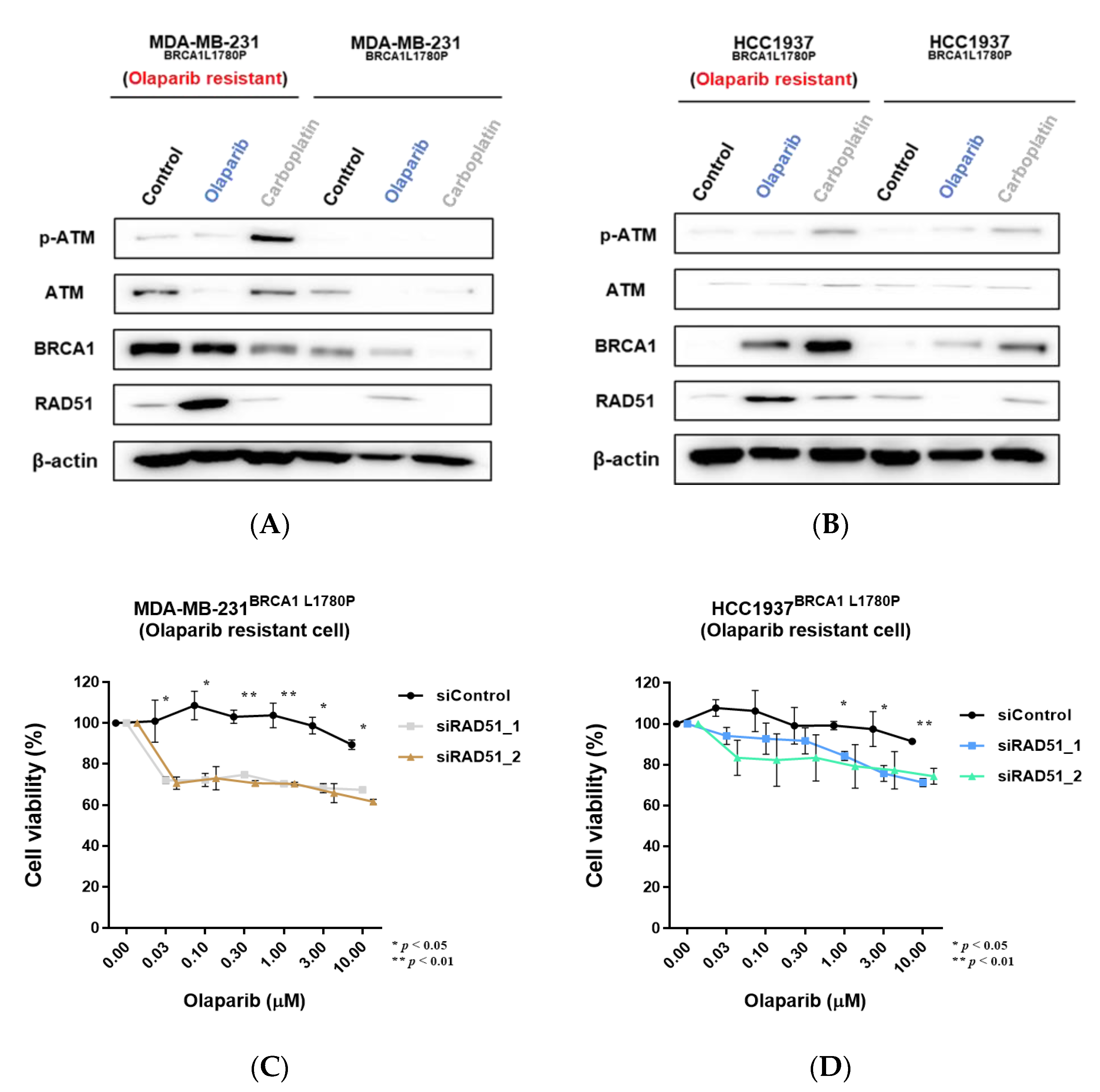
3.4. Effect of BRCA1 L1780P Mutation in RAD51 Induction upon Olaparib and Carboplatin Treatment
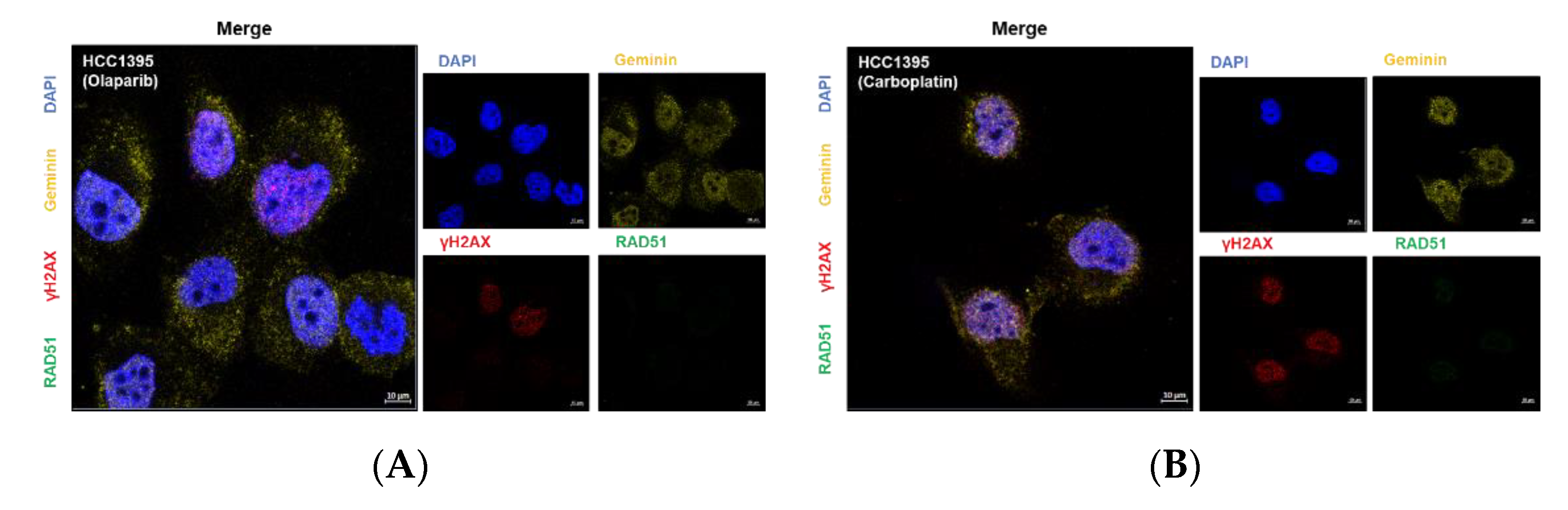
3.5. Nuclear RAD51 Recruitment Is Decreased in BRCA1 L1780P Mutated Cells
3.6. Olaparib Sensitivity Induced by Downregulation of RAD51 in BRCA-Proficient Cells
3.7. Downregulation of RAD51 in Olaparib-Resistant Cell Lines Restored Olaparib Sensitivity
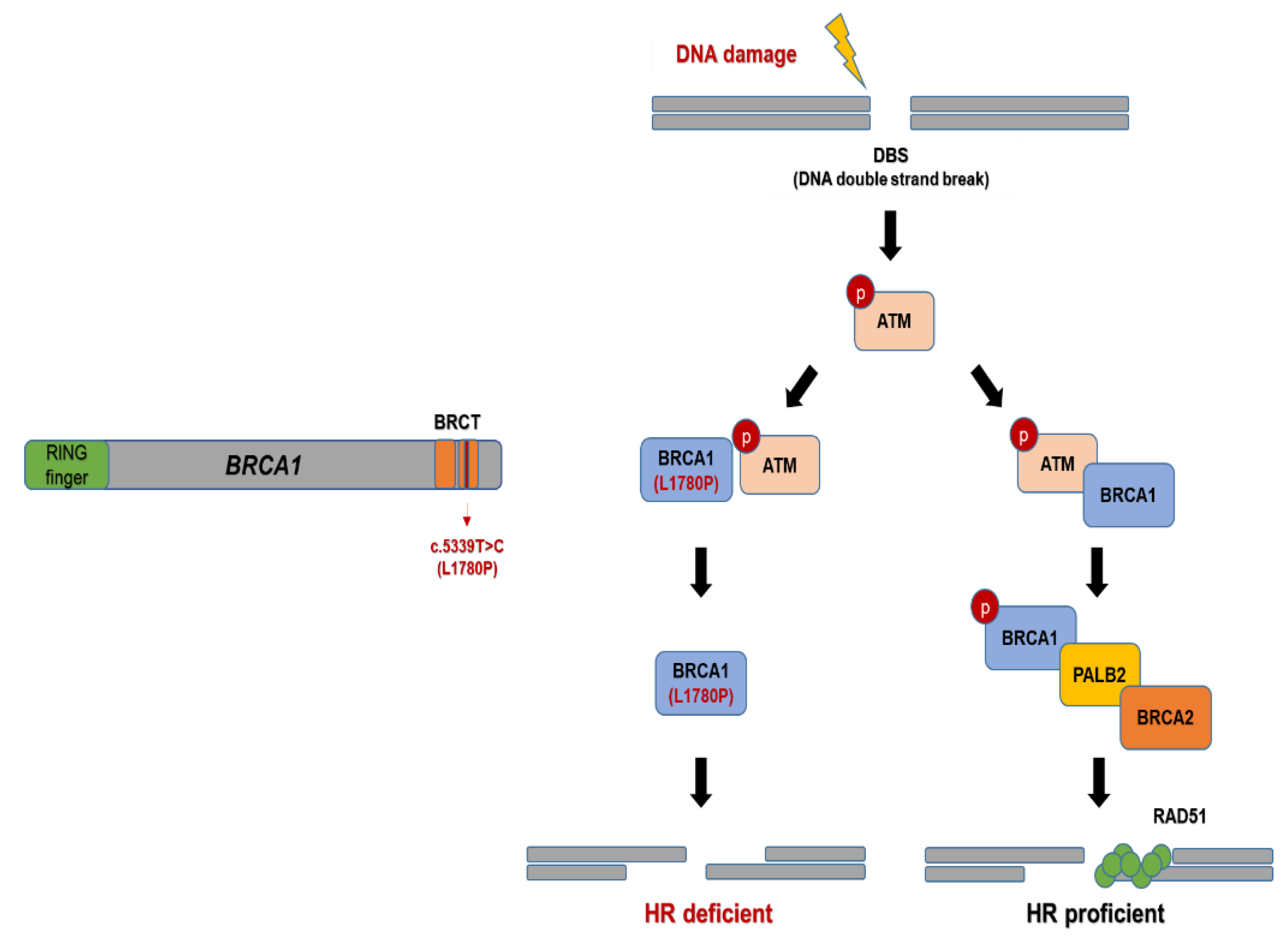
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Ryu, W.J.; Sohn, J.H. Molecular Targets and Promising Therapeutics of Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1008. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef]
- Rosenberg, S.M.; Ruddy, K.J.; Tamimi, R.M.; Gelber, S.; Schapira, L.; Come, S.; Borges, V.F.; Larsen, B.; Garber, J.E.; Partridge, A.H. BRCA1 and BRCA2 Mutation Testing in Young Women With Breast Cancer. JAMA Oncol. 2016, 2, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Trainer, A.H.; Lewis, C.R.; Tucker, K.; Meiser, B.; Friedlander, M.; Ward, R.L. The role of BRCA mutation testing in determining breast cancer therapy. Nat. Rev. Clin. Oncol. 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev. Pathol. 2009, 4, 461–487. [Google Scholar] [CrossRef]
- Paull, T.T.; Lee, J.H. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 2005, 4, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef]
- Sung, P.; Krejci, L.; Van Komen, S.; Sehorn, M.G. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003, 278, 42729–42732. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Connor, A.A.; Gallinger, S. Characterization, Detection, and Treatment Approaches for Homologous Recombination Deficiency in Cancer. Trends Mol. Med. 2017, 23, 1121–1137. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Hayes, F.; Cayanan, C.; Barilla, D.; Monteiro, A.N. Functional assay for BRCA1: Mutagenesis of the COOH-terminal region reveals critical residues for transcription activation. Cancer Res. 2000, 60, 2411–2418. [Google Scholar]
- Williams, R.S.; Lee, M.S.; Hau, D.D.; Glover, J.N. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat. Struct. Mol. Biol. 2004, 11, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- den Brok, W.D.; Schrader, K.A.; Sun, S.; Tinker, A.V.; Zhao, E.Y.; Aparicio, S.; Gelmon, K.A. Homologous Recombination Deficiency in Breast Cancer: A Clinical Review. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.; et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010, 70, 4880–4890. [Google Scholar] [CrossRef]
- Park, J.S.; Nam, E.J.; Park, H.S.; Han, J.W.; Lee, J.Y.; Kim, J.; Kim, T.I.; Lee, S.T. Identification of a Novel BRCA1 Pathogenic Mutation in Korean Patients Following Reclassification of BRCA1 and BRCA2 Variants According to the ACMG Standards and Guidelines Using Relevant Ethnic Controls. Cancer Res. Treat. 2017, 49, 1012–1021. [Google Scholar] [CrossRef]
- Park, H.S.; Ryu, J.M.; Park, J.S.; Im, S.A.; Jung, S.Y.; Kim, E.K.; Park, W.C.; Min, J.W.; Lee, J.; You, J.Y.; et al. Clinicopathological Features of Patients with the BRCA1 c.5339T>C (p.Leu1780Pro) Variant. Cancer Res. Treat. 2020, 52, 680–688. [Google Scholar] [CrossRef]
- Ryu, J.M.; Kang, G.; Nam, S.J.; Kim, S.W.; Yu, J.; Lee, S.K.; Bae, S.Y.; Park, S.; Paik, H.J.; Kim, J.W.; et al. Suggestion of BRCA1 c.5339T>C (p.L1780P) variant confer from ‘unknown significance’ to ‘Likely pathogenic’ based on clinical evidence in Korea. Breast 2017, 33, 109–116. [Google Scholar] [CrossRef]
- Yoon, K.A.; Park, B.; Lee, B.I.; Yang, M.J.; Kong, S.Y.; Lee, E.S. Clinically Significant Unclassified Variants in BRCA1 and BRCA2 genes among Korean Breast Cancer Patients. Cancer Res. Treat. 2017, 49, 627–634. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Smith, S.E.; Mellor, P.; Ward, A.K.; Kendall, S.; McDonald, M.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Napper, S.; Anderson, D.H. Molecular characterization of breast cancer cell lines through multiple omic approaches. Breast Cancer Res. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Furuta, S.; Jiang, X.; Gu, B.; Cheng, E.; Chen, P.L.; Lee, W.H. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9176–9181. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wagner, K.U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rajan, J.V.; Wang, M.; Marquis, S.T.; Chodosh, L.A. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 13078–13083. [Google Scholar] [CrossRef]
- Miklikova, S.; Trnkova, L.; Plava, J.; Bohac, M.; Kuniakova, M.; Cihova, M. The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer. Cancers 2021, 13, 575. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Spirek, M.; Chaurasiya, K.R.; Ward, J.D.; Carzaniga, R.; Yu, X.; Egelman, E.H.; Collinson, L.M.; Rueda, D.; Krejci, L.; et al. Rad51 Paralogs Remodel Pre-synaptic Rad51 Filaments to Stimulate Homologous Recombination. Cell 2015, 162, 271–286. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutierrez-Enriquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutierrez-Enriquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzman, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Goessl, C.; Domchek, S. Olaparib for Metastatic Germline BRCA-Mutated Breast Cancer. N. Engl. J. Med. 2017, 377, 1792–1793. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Janysek, D.C.; Kim, J.; Duijf, P.H.G.; Dray, E. Clinical use and mechanisms of resistance for PARP inhibitors in homologous recombination-deficient cancers. Transl. Oncol. 2021, 14, 101012. [Google Scholar] [CrossRef]
- Kim, D.S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Martin, R.W.; Orelli, B.J.; Yamazoe, M.; Minn, A.J.; Takeda, S.; Bishop, D.K. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007, 67, 9658–9665. [Google Scholar] [CrossRef]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.D.; Ryu, W.-J.; Han, H.J.; Kim, T.Y.; Kim, M.H.; Sohn, J. Molecular Characterization of BRCA1 c.5339T>C Missense Mutation in DNA Damage Response of Triple-Negative Breast Cancer. Cancers 2022, 14, 2405. https://doi.org/10.3390/cancers14102405
Lee JD, Ryu W-J, Han HJ, Kim TY, Kim MH, Sohn J. Molecular Characterization of BRCA1 c.5339T>C Missense Mutation in DNA Damage Response of Triple-Negative Breast Cancer. Cancers. 2022; 14(10):2405. https://doi.org/10.3390/cancers14102405
Chicago/Turabian StyleLee, Jeong Dong, Won-Ji Ryu, Hyun Ju Han, Tae Yeong Kim, Min Hwan Kim, and Joohyuk Sohn. 2022. "Molecular Characterization of BRCA1 c.5339T>C Missense Mutation in DNA Damage Response of Triple-Negative Breast Cancer" Cancers 14, no. 10: 2405. https://doi.org/10.3390/cancers14102405