
Epidemiology of Glioblastoma Multiforme–Literature Review

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
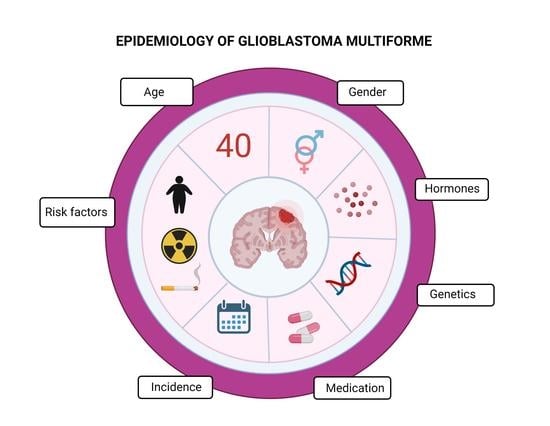
1. Introduction
2. Classification of GBM
- Glioblastoma, isocitrate dehydrogenase (IDH) wildtype (90% of cases), developing de novo at about 60 years of age;
- Glioblastoma, IDH-mutant (10% of cases), secondary GBM that usually develops in younger patients with gliomas of higher differentiation (WHO grades I-III); it carries a significantly better prognosis than wildtype IDH [16];
- Glioblastoma not otherwise specified (NOS), the IDH mutation status could not be determined due to a lack of histological or molecular material for testing;
- Not-elsewhere-classified (NEC) Glioblastoma, fourth category distinguished in recent years. The necessary determinations regarding the classification of the tumor have been made, but the results do not allow matching the tumor to any of the aforementioned categories of the 2016 WHO division. This situation may occur in the case of discrepancies between the clinical, histological, immunohistological, and genetic features of the tumor. There is also the possibility that a GBM subunit exists with an unknown combination of features that is not yet classified in the WHO division.
3. Pathogenesis of GBM
3.1. GBM Site in the Brain
3.2. Genetic Pathogenesis
- ATRX (a-thalassemia/mental-retardation-syndrome-X-linked)mutation. The ATRX gene located on Xq21.1 encodes a protein involved in the chromatin-rearrangement pathway, allowing histone H3.3 to be incorporated into heterochromatin [33]. ATRX mutations occur in approximately 57% of secondary GBM; in GBM cells, ATRX mutations occur more frequently in IDH-mutant GBM than in wildtype (71% vs. exceptions) [15] and co-occur with IDH1 and TP53 mutations [34,35]. ATRX mutations are positive prognostic factors [36,37]. In a prospective study conducted on a cohort of patients with astrocytic tumors (grade I-IV), those with a loss of ATRX expression had a better prognosis than those who retained ATRX expression and a co-occurring IDH mutation [38].
- TERT (Telomerase Reverse Transcriptase) promoter mutation. The TERT gene encodes telomerase, an enzyme responsible for adding the missing 3′ end of a DNA strand during replication. The mutation of the TERT gene promoter results in increased telomerase activity and telomere elongation, suggesting that maintaining the presence of telomeres is essential for brain tumor formation [39,40]. The two most common mutations of the TERT promoter are C228T and C250T, located at base pairs 124 and 146, respectively, which encode this promoter [41]. These mutations can lead to up to a fourfold increase in TERT expression [39,42]. TERT mutations are present in up to 80% of GBM [39,43,44,45,46]. The role of TERT promoter mutation as a prognostic factor has not been unequivocally determined due to numerous confounding co-occurring factors (age, surgical intervention, IDH, EGFR mutations, and MGMT methylation status) [47]. TERT promoter mutations occur more frequently in IDH-wildtype GBM than IDH-mutant GBM (72% vs. 26%) [15]. Further prospective studies are needed on large cohorts of a homogeneous patient population (for example, IDH-wildtype and O6-methylguanine DNA methyltransferase (MGMT) promoter-unmethylated glioma) to independently assess the prognostic impact of TERT promoter mutations [47].
- TP53 (Tumor protein P53)mutation: The TP53 gene is located on human chromosome 17p13.1. The functional p53 protein is a homotetramer that plays a key role in the regulatory network, controlling proliferation, survival, genome integrity, and other cellular functions. The presence of TP53 mutations is associated with the progression of GBM [48]. The inactivation of p53 correlates with increased invasiveness [49], decreased cell apoptosis [50], and increased proliferation [51]. Cell lines carrying the p53-inactivating mutation show greater resistance to DNA-damaging chemotherapeutics, such as cisplatin [50]. Although TP53 mutations correlate with poor prognoses in other cancers [52], they have no prognostic value in GBM [51,53,54]. TP53 mutations are more common in IDH-mutant GBM than IDH-wildtype GBM (81% vs. 27%) [15]. The gain of function (GOF) TP53 mutation results in a new function or altered expression; in GBM, it leads to increased malignancy of cells by increasing their proliferation, migration, invasion, metastasis, drug resistance, and genome instability, and increasing survival [55,56,57,58]. Wang, Xiang et al. [59] reported that GOF mutations are associated with worse OS, and that they reduce GBM sensitivity to temozolomide by increasing MGMT expression [59].
- B-RAF V600E mutation: B-RAF is part of the RAS-RAF-MEK-ERK MAP kinase pathway. This precisely regulated pathway is responsible for cell growth; mutations that confer the constitutive B-RAF kinase activity would result in uncontrolled cell proliferation and tumor formation. The V600E mutation involves the substitution of valine for glutamate at position 600 of the B-RAF protein, producing the permanently activated serine/threonine kinase B-RAF, which activates extracellular signal-regulated kinase 1 and 2 (ERK1/2) and other mitogen-activated protein (MAP) kinases. In the literature, the frequency of all B-RAF mutations in GBM is estimated at 2–6%. In one study [60], patients with GBM from four studies were evaluated, and 8 out of 505 (1.5%) showed the presence of the B-RAF V600E mutation. The mutation may be a convenient point of entry for effective personalized anticancer therapy with kinase inhibitors, as evidenced by published case reports, such as the clinical response to vemurafenib (a B-RAF kinase inhibitor) in three pediatric high-grade gliomas [61].
- GATA4 (GATA-binding protein 4): GATA4 is a transcription factor of the GATA6 family, considered a suppressor gene. In normal astrocytes, GATA4 does not affect cell growth; however, in mice with knockout GATA4 genes and null-p53 status, the absence of GATA4 induces a transformation associated with increased proliferation, resistance to chemotherapy, and radiotherapy-induced apoptosis [62]. A series of studies conducted by Agnihotri et al. [63] showed that: (i) in 94/163 human GBM tumor cells, GATA4 expression was lost, (ii) GATA4 inhibited transformation to GBM in vitro and in vivo, and (iii) the re-expression of GATA4 in GBM cells sensitized them to telosomide, regardless of the MGMT mutation status. The role of GATA4 in response to telosomide suggests the utility of the GATA4 mutation status as a predictive biomarker, and this needs to be confirmed in further studies.
- FGFR1 (Fibroblast Growth Factor Receptor (1)): the FGFR family of proteins is a group of transmembrane receptors with tyrosine kinase function. The exact impact of signaling by FGFR on the pathobiological aspects of individual cancers remains unknown [64]. The strongest evidence suggests that FGFR1 contributes to poor prognosis in GBM, and signaling through this pathway is associated with increased radioresistance, invasiveness, and stemness [65,66,67].
- EGFR (epidermal growth factor receptor): EGFR is a receptor with tyrosine kinase activity that is activated by EGF (epidermal growth factor). EGFR promotes cell proliferation by activating the MAPK and PI3K-Akt pathways [68]. The EGFR gene is located at locus 7p12 and its amplification is observed in approximately 40% of GBM cases [69]. The amplification of EGFR has been associated with poor prognoses by some authors, but the results are inconclusive [70,71,72,73,74]. EGFR amplification is more common in IDH-wildtype GBM than IDH-mutant GBM (35% vs. exceptional) [15]. The most common EGFR mutation is variant-III EGFR mutation (EGFRvIII), involving a deletion without a shift of the reading frame of base pair 801 extending from exon 2 to 7, and it has attracted much research interest. This mutation leads to the constitutive activation of EGFR, resulting in the activation of downstream tyrosine kinase pathways. EGFRvIII mutation occurs almost exclusively in the presence of EGFR amplification [75]. Attempts to create a vaccine targeting EGFRvIII (Rindopepimut) ended in the phase-IV clinical trial in 2016 due to a lack of improvement in OS.
- MGMT (O6-methylguanine DNA methyltransferase): the MGMT gene is located on chromosome 10q26 and encodes a protein responsible for DNA repair, removing an alkyl group from the O6 position of guanine, an important DNA alkylation site. The presence of MGMT promoter methylation is a positive predictor of better OS. The authors of a study published in NEJM [76] suggested the usefulness of determining the MGMT promoter methylation status by methylation-specific PCR in order to identify patients who may benefit from including temozolomide with standard radiotherapy compared with radiotherapy alone. Temozolomide works by methylating DNA at the N7 and O6 atoms for guanine and N3 for adenine. Methylation sites can be repaired by specialized enzymes, such as MGMT. In a paper published in 2012 [77], the efficacy of temozolomide was greater for a methylated MGMT promoter in GBM cells (i.e., reduced MGMT expression). The use of the MGMT inhibitor O6-benzylguanine (O6-BG) restored temozolomide (TMZ) sensitivity to TMZ-resistant cell lines LN-18 and T98G [78].
- WT1 (The Wilms tumor gene): WT1 was first identified as the gene responsible for the development of the Wilms kidney tumor that primarily affects children. The WT1 gene is located at locus 11p13 and functions as a zinc finger-like transcription factor. Despite the initial classification of WT1 as a suppressor gene, the overexpression of WT-1 in many cancers (breast cancer and acute leukemias) [79,80] has led to its recognition as an oncogene [81]. In a study conducted in 2004 [41], 48 out of 51 GBM samples (94%) showed positive staining for the WT-1 protein.
- PTEN (Phosphatase and tensin homolog): the PTEN gene is a suppressor gene located on 10q23. LOH (loss of heterozygosity) or methylation mutation disrupt the pathways that use phosphatidylinositol 3-kinase (PI3K) and are found in at least 60% of GBM cases [82]. Loss of PTEN function due to mutation or loss of heterozygosity (LOH) is associated with poor prognosis of GBM. PTEN is a protein with protein phosphatase and lipid phosphatase functions, and most of the onco-suppressive properties are due to the lipid phosphatase properties [83]. The PI3K/Akt pathway is blocked by PTEN, and loss of functional PTEN impairs the regulation of cell survival, cell growth, and proliferation [84]. According to Koul [82], loss of PTEN expression is indicative of the progression of a highly malignant tumor—PTEN is present in most low-grade tumors. Brito et al. [26] reported that PTEN deletion in IDH-wildtype GBM is associated with better OS.
4. Survival and Prognostic Factors
4.1. Incidence
4.2. Age
4.3. Survival
4.4. Urban/Rural Socioeconomic Status
5. Protective Factors
5.1. Gender and Hormones
5.2. Non-Steroidal Anti-Inflammatory Drugs and Paracetamol
5.3. Other Medications
5.3.1. Antihistamines
5.3.2. Statins
5.3.3. Cannabinoids
- Apoptosis and cytotoxic autophagy;
- Mechanisms that inhibit cell proliferation;
- Anti-angiogenic mechanisms.
5.3.4. Atopy
6. Risk Factors
6.1. Tobacco Smoking and Nitrosamines
6.2. Race/Ethnicity
6.3. Ionizing Radiation
6.4. Head Injury
6.5. Obesity
6.6. Growth
6.7. Metals
6.8. Nutritional Factors, Chemicals, and Pesticides
6.9. Coffee and Tea
6.10. Alcohol Use
6.11. Sleep and Melatonin
6.12. Inflammation
6.13. Electromagnetic Radiation
7. Treatment of GBM
8. Conclusions
9. Limitation
Author Contributions
Funding
Conflicts of Interest
References
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, B.; Rosenthal, M.A. Survival comparison between glioblastoma multiforme and other incurable cancers. J. Clin. Neurosci. 2010, 17, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Wesseling, P. Histologic classification of gliomas. Handb. Clin. Neurol. 2016, 134, 71–95. [Google Scholar] [PubMed]
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Fabbro-Peray, P.; Zouaoui, S.; Darlix, A.; Fabbro, M.; Pallud, J.; Rigau, V.; Mathieu-Daude, H.; Bessaoud, F.; Bauchet, F.; Riondel, A.; et al. Association of patterns of care, prognostic factors, and use of radiotherapy–temozolomide therapy with survival in patients with newly diagnosed glioblastoma: A French national population-based study. J. Neurooncol. 2019, 142, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Herrera, F.; Castro-Sierra, E.; Gordillo-Domínguez, L.F.; Vaca-Ruiz, M.A.; Santana-Montero, B.; Perezpeña-Diazconti, M.; González-Carranza, V.; Torres-García, S.; Chico-Ponce De León, F. Glioblastoma multiforme in children: Experience at Hospital Infantil de Mexico Federico Gomez. Child’s Nerv. Syst. 2009, 25, 551–557. [Google Scholar] [CrossRef]
- Perkins, S.M.; Rubin, J.B.; Leonard, J.R.; Smyth, M.D.; El Naqa, I.; Michalski, J.M.; Simpson, J.R.; Limbrick, D.L.; Park, T.S.; Mansur, D.B. Glioblastoma in children: A single-institution experience. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 1117–1121. [Google Scholar] [CrossRef]
- Das, K.K.; Mehrotra, A.; Nair, A.P.; Kumar, S.; Srivastava, A.K.; Sahu, R.N.; Kumar, R. Pediatric glioblastoma: Clinico-radiological profile and factors affecting the outcome. Child’s Nerv. Syst. 2012, 28, 2055–2062. [Google Scholar] [CrossRef]
- Suri, V.; Das, P.; Jain, A.; Sharma, M.C.; Borkar, S.A.; Suri, A.; Gupta, D.; Sarkar, C. Pediatric glioblastomas: A histopathological and molecular genetic study. Neuro-Oncology 2009, 11, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Faury, D.; Nantel, A.; Dunn, S.E.; Guiot, M.C.; Haque, T.; Hauser, P.; Garami, M.; Bognár, L.; Hanzély, Z.; Liberski, P.P.; et al. Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J. Clin. Oncol. 2007, 25, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, G.J.; Farwell, J.R.; Flannery, J.T. Astrocytomas in childhood: A population-based study. Surg. Neurol. 1985, 23, 64–68. [Google Scholar] [CrossRef]
- Das, K.K.; Kumar, R. Pediatric Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 297–312. [Google Scholar]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro-Oncology 2020, 22, IV1–IV96. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef]
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol. 2018, 22, 215–222. [Google Scholar] [CrossRef]
- Fu, Y.; Zheng, S.; Zheng, Y.; Huang, R.; An, N.; Liang, A.; Hu, C. Glioma derived isocitrate dehydrogenase-2 mutations induced up-regulation of HIF-1α and β-catenin signaling: Possible impact on glioma cell metastasis and chemo-resistance. Int. J. Biochem. Cell Biol. 2012, 44, 770–775. [Google Scholar] [CrossRef]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [Green Version]
- Tandel, G.S.; Biswas, M.; Kakde, O.G.; Tiwari, A.; Suri, H.S.; Turk, M.; Laird, J.R.; Asare, C.K.; Ankrah, A.A.; Khanna, N.N.; et al. A review on a deep learning perspective in brain cancer classification. Cancers 2019, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Aldape, K.; Brat, D.J.; Capper, D.; Ellison, D.W.; Hawkins, C.; Paulus, W.; Perry, A.; Reifenberger, G.; Figarella-Branger, D.; et al. Announcing cIMPACT-NOW: The Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy. Acta Neuropathol. 2017, 133, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Lin, Y.; Zinn, P.; Su, J.; Pan, I.W. Patient and treatment factors associated with survival among pediatric glioblastoma patients: A Surveillance, Epidemiology, and End Results study. J. Clin. Neurosci. 2018, 47, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Shubham, S.; Mandal, K.; Trivedi, V.; Chauhan, R.; Naseera, S. Survival and prognostic factors for glioblastoma multiforme: Retrospective single-institutional study. Indian J. Cancer 2017, 54, 362–367. [Google Scholar]
- Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Bravo Marques, J.M.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968. [Google Scholar] [CrossRef] [Green Version]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain tumours: Rise in glioblastoma multiforme incidence in England 1995–2015 suggests an adverse environmental or lifestyle factor. Int. J. Environ. Res. Public Health 2018, 2018, 7910754. [Google Scholar]
- Bohn, A.; Braley, A.; De La Vega, P.R.; Carlos Zevallos, J.; Barengo, N.C. The association between race and survival in glioblastoma patients in the US: A retrospective cohort study. PLoS ONE 2018, 13, e0198581. [Google Scholar] [CrossRef]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, I.; Cockburn, M.; Cozen, W.; Wang, Y.P.; Preston-Martin, S. A population-based description of glioblastoma multiforme in Los Angeles County, 1974–1999. Cancer 2005, 104, 2798–2806. [Google Scholar] [CrossRef]
- Hansen, S.; Rasmussen, B.K.; Laursen, R.J.; Kosteljanetz, M.; Schultz, H.; Nørgård, B.M.; Guldberg, R.; Gradel, K.O. Treatment and survival of glioblastoma patients in Denmark: The Danish Neuro-Oncology Registry 2009–2014. J. Neurooncol. 2018, 139, 479–489. [Google Scholar] [CrossRef]
- Li, K.; Lu, D.; Guo, Y.; Wang, C.; Liu, X.; Liu, Y.; Liu, D. Trends and patterns of incidence of diffuse glioma in adults in the United States, 1973–2014. Cancer Med. 2018, 7, 5281–5290. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Gerges, N.; Korshunov, A.; Sabha, N.; Khuong-Quang, D.A.; Fontebasso, A.M.; Fleming, A.; Hadjadj, D.; Schwartzentruber, J.; Majewski, J.; et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012, 124, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, A.; Park, S.H.; Seo, J.W.; Park, C.K. Immunohistochemical analysis of ATRX, IDH1 and p53 in glioblastoma and their correlations with patient survival. J. Korean Med. Sci. 2016, 31, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, C.; Zhang, W.; Wang, G.; Yao, K.; Wang, Z.; Li, G.; Qian, Z.; Li, Y.; Jiang, T.; et al. ATRX, IDH1-R132H and Ki-67 immunohistochemistry as a classification scheme for astrocytic tumors. Oncoscience 2016, 3, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; Von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinagre, J.; Pinto, V.; Celestino, R.; Reis, M.; Pópulo, H.; Boaventura, P.; Melo, M.; Catarino, T.; Lima, J.; Lopes, J.M.; et al. Telomerase promoter mutations in cancer: An emerging molecular biomarker? Virchows Arch. 2014, 465, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, Y.; Okamoto, H.; Mineta, T.; Tabuchi, K. Expression of the Wilms’ tumor gene product WT1 in glioblastomas and medulloblastomas. Brain Tumor Pathol. 2004, 21, 113–116. [Google Scholar] [CrossRef]
- Fredriksson, N.J.; Ny, L.; Nilsson, J.A.; Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014, 46, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegl-Kreinecker, S.; Lötsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro-Oncology 2015, 17, 1231–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosrati, M.A.; Malmström, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.L.; Bratthäll, C.; Strandéus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663–16673. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Qi, C.; Maling, G.; Xiang, W.; Yanhui, L.; Ruofei, L.; Yunhe, M.; Jiewen, L.; Qing, M. TERT mutation in glioma: Frequency, prognosis and risk. J. Clin. Neurosci. 2016, 26, 57–62. [Google Scholar] [CrossRef]
- Olympios, N.; Gilard, V.; Marguet, F.; Clatot, F.; Di Fiore, F.; Fontanilles, M. TERT promoter alterations in glioblastoma: A systematic review. Cancers 2021, 13, 1147. [Google Scholar] [CrossRef]
- Wang, T.J.; Huang, M.S.; Hong, C.Y.; Tse, V.; Silverberg, G.D.; Hsiao, M. Comparisons of tumor suppressor p53, p21, and p16 gene therapy effects on glioblastoma tumorigenicity in Situ. Biochem. Biophys. Res. Commun. 2001, 287, 173–180. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Moon, S.I.; Yoo, D.H.; Lee, H.C.; Park, I.C.; Rhee, C.H.; Hong, S. Il Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int. J. Oncol. 2006, 28, 119–125. [Google Scholar]
- England, B.; Huang, T.; Karsy, M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumor Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, J.A.; Glesmann, N.; Beck, M.; Krex, D.; Klockgether, T.; Schackert, G.; Schlegel, U. Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2000, 48, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Hans, C.; Jones, B.; Iversen, E.S.; McLendon, R.E.; Rasheed, B.K.A.; Dobra, A.; Dressman, H.K.; Bigner, D.D.; Nevins, J.R.; et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res. 2005, 65, 4051–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad Complex Opposes p63 to Empower TGFβ-Induced Metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-function (GOF) mutant p53 as actionable therapeutic target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.X.; Liu, J.P.; You, C.; Liu, Y.H.; Mao, Q. Gain of function of mutant TP53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Bautista, F.; Paci, A.; Minard-Colin, V.; Dufour, C.; Grill, J.; Lacroix, L.; Varlet, P.; Valteau-Couanet, D.; Geoerger, B. Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr. Blood Cancer 2014, 61, 1101–1103. [Google Scholar] [CrossRef]
- Agnihotri, S.; Wolf, A.; Picard, D.; Hawkins, C.; Guha, A. GATA4 is a regulator of astrocyte cell proliferation and apoptosis in the human and murine central nervous system. Oncogene 2009, 28, 3033–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, S.; Wolf, A.; Munoz, D.M.; Smith, C.J.; Gajadhar, A.; Restrepo, A.; Clarke, I.D.; Fuller, G.N.; Kesari, S.; Dirks, P.B.; et al. A GATA4-regulated tumor suppressor network represses formation of malignant human astrocytomas. J. Exp. Med. 2011, 208, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.S.; Jimenez-Pascual, A.; Kordowski, A.; Pugh, J.; Rao, S.; Silver, D.J.; Alban, T.; Watson, D.B.; Chen, R.; McIntyre, T.M.; et al. ADAMDEC1 maintains a novel growth factor signaling loop in cancer stem cells. bioRxiv 2019, 531509. [Google Scholar] [CrossRef] [Green Version]
- Gouazé-Andersson, V.; Delmas, C.; Taurand, M.; Martinez-Gala, J.; Evrard, S.; Mazoyer, S.; Toulas, C.; Cohen-Jonathan-moyal, E. FGFR1 induces glioblastoma radioresistance through the PLCg/Hif1a pathway. Cancer Res. 2016, 76, 3036–3044. [Google Scholar] [CrossRef] [Green Version]
- Kowalski-Chauvel, A.; Gouaze-Andersson, V.; Baricault, L.; Martin, E.; Delmas, C.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. Alpha6-integrin regulates FGFR1 expression through the ZEB1/YAP1 transcription complex in glioblastoma stem cells resulting in enhanced proliferation and stemness. Cancers 2019, 11, 406. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Leung, H.Y. Targeting the EGFR-family for Therapy: Biological Challenges and Clinical Perspective. Curr. Pharm. Des. 2012, 18, 2672–2679. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, E.W.; Cohen, H.; Lee, S.R.; Bhalla, S.K.; Bloom, J.; Hayes, R.L.; Miller, D.C. Survival of patients with glioblastoma multiforme is not influenced by altered expression of p16, p53, EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 1998, 8, 655–667. [Google Scholar] [CrossRef]
- Cowppli-Bony, A.; Bouvier, G.; Rué, M.; Loiseau, H.; Vital, A.; Lebailly, P.; Fabbro-Peray, P.; Baldi, I. Brain tumors and hormonal factors: Review of the epidemiological literature. Cancer Causes Control 2011, 22, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Tachibana, I.; Passe, S.M.; Huntley, B.K.; Borell, T.J.; Iturria, N.; O’Fallon, J.R.; Schaefer, P.L.; Scheithauer, B.W.; James, C.D.; et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J. Natl. Cancer Inst. 2001, 93, 1246–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, M.L.; Lamborn, K.R.; Takahashi, M.; Chen, P.; Israel, M.A.; Berger, M.S.; Godfrey, T.; Nigro, J.; Prados, M.; Chang, S.; et al. Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res. 2001, 61, 1122–1128. [Google Scholar] [PubMed]
- Heimberger, A.B.; Sampson, J.H. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin. Biol. Ther. 2009, 9, 1087–1098. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Kanzawa, T.; Bedwell, J.; Kondo, Y.; Kondo, S.; Germano, I.M. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J. Neurosurg. 2003, 99, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, Y.; Ando, A.; Egawa, C.; Taguchi, T.; Tamaki, Y.; Tamaki, H.; Sugiyama, H.; Noguchi, S. High expression of Wilms’ tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin. Cancer Res. 2002, 8, 1167–1171. [Google Scholar]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Nakagawa, M.; Yamagami, T.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; Nasu, K.; et al. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994, 84, 3071–3079. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Ware, J.L.; Chen, M.Y.; Graf, M.R.; Van Meter, T.E.; Dos Santos, W.G.; Fillmore, H.L.; Broaddus, W.C. Effect of WT1 gene silencing on the tumorigenicity of human glioblastoma multiforme cells: Laboratory investigation. J. Neurosurg. 2010, 112, 18–25. [Google Scholar] [CrossRef]
- Koul, D. PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Knobbe, C.B.; Merlo, A.; Reifenberger, G. Pten signalling in gliomas. Neuro-Oncology 2002, 4, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Ma, W.; Chen, Y.; Yu, Y.; Zhu, D.; Shi, J.; Zhang, Y. Impact of gender on the survival of patients with glioblastoma. Biosci. Rep. 2018, 38, BSR20180752. [Google Scholar] [CrossRef] [Green Version]
- Dho, Y.-S.; Jung, K.-W.; Ha, J.; Seo, Y.; Park, C.-K.; Won, Y.-J.; Yoo, H. An Updated Nationwide Epidemiology of Primary Brain Tumors in Republic of Korea, 2013. Brain Tumor Res. Treat. 2017, 5, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobec-Meić, B.; Pikija, S.; Cvetko, D.; Trkulja, V.; Pažanin, L.; Kudelić, N.; Rotim, K.; Pavliček, I.; Kostanjevec, R. Intracranial tumors in adult population of the Varaždin County (Croatia) 1996–2004: A population-based retrospective incidence study. J. Neurooncol. 2006, 78, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Dobes, M.; Khurana, V.G.; Shadbolt, B.; Jain, S.; Smith, S.F.; Smee, R.; Dexter, M.; Cook, R. Increasing incidence of glioblastoma multiforme and meningioma, and decreasing incidence of Schwannoma (2000-2008): Findings of a multicenter Australian study. Surg. Neurol. Int. 2011, 2, 176. [Google Scholar]
- Fleury, A.; Menegoz, F.; Grosclaude, P.; Daures, J.P.; Henry-Amar, M.; Raverdy, N.; Schaffer, P.; Poisson, M.; Delattre, J.Y. Descriptive epidemiology of cerebral gliomas in France. Cancer 1997, 79, 1195–1202. [Google Scholar] [CrossRef]
- Fuentes-Raspall, R.; Vilardell, L.; Perez-Bueno, F.; Joly, C.; Garcia-Gil, M.; Garcia-Velasco, A.; Marcos-Gragera, R. Population-based incidence and survival of central nervous system (CNS) malignancies in Girona (Spain) 1994–2005. J. Neurooncol. 2011, 101, 117–123. [Google Scholar] [CrossRef]
- Fuentes-Raspall, R.; Solans, M.; Roca-Barceló, A.; Vilardell, L.; Puigdemont, M.; Del Barco, S.; Comas, R.; García-Velasco, A.; Astudillo, A.; Carmona-Garcia, M.C.; et al. Descriptive epidemiology of primary malignant and non-malignant central nervous tumors in Spain: Results from the Girona Cancer Registry (1994–2013). Cancer Epidemiol. 2017, 50, 1–8. [Google Scholar] [CrossRef]
- Gittleman, H.; Boscia, A.; Ostrom, Q.T.; Truitt, G.; Fritz, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Survivorship in adults with malignant brain and other central nervous system tumor from 2000–2014. Neuro-Oncology 2018, 20, VII6–VII16. [Google Scholar] [CrossRef] [PubMed]
- Gousias, K.; Markou, M.; Voulgaris, S.; Goussia, A.; Voulgari, P.; Bai, M.; Polyzoidis, K.; Kyritsis, A.; Alamanos, Y. Descriptive epidemiology of cerebral gliomas in Northwest Greece and study of potential predisposing factors, 2005–2007. Neuroepidemiology 2009, 33, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.K.Y.; Reijneveld, J.C.; Enting, R.H.; Bienfait, H.P.; Robe, P.; Baumert, B.G.; Visser, O. Changing incidence and improved survival of gliomas. Eur. J. Cancer 2014, 50, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, S.B.; Rahimi-Movaghar, V.; Shokraneh, F.; Saadat, S.; Ramezani, R. Epidemiology of primary CNS tumors in Iran: A systematic review. Asian Pac. J. Cancer Prev. 2013, 14, 3979–3985. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.-W.; Ha, J.; Lee, S.H.; Won, Y.-J.; Yoo, H. An Updated Nationwide Epidemiology of Primary Brain Tumors in Republic of Korea. Brain Tumor Res. Treat. 2013, 1, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Larjavaara, S.; Mäntylä, R.; Salminen, T.; Haapasalo, H.; Raitanen, J.; Jääskeläinen, J.; Auvinen, A. Incidence of gliomas by anatomic location. Neuro-Oncology 2007, 9, 319–325. [Google Scholar] [CrossRef]
- Natukka, T.; Raitanen, J.; Haapasalo, H.; Auvinen, A. Incidence trends of adult malignant brain tumors in Finland, 1990–2016. Acta Oncol. 2019, 58, 990–996. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology 2017, 19, v1–v88. [Google Scholar] [CrossRef] [Green Version]
- Schoenberg, B.S.; Christine, B.W.; Whisnant, J.P. The descriptive epidemiology of primary intracranial neoplasms: The connecticut experience. Am. J. Epidemiol. 1976, 104, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.V.; Davis, F.G.; Shaw, A.; Louchini, R.; Shack, L.; Woods, R.; Kruchko, C.; Spinelli, J.; Guiot, M.C.; Perry, J.; et al. Malignant primary brain and other central nervous system tumors diagnosed in Canada from 2009 to 2013. Neuro-Oncology 2019, 21, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Wanner, M.; Rohrmann, S.; Korol, D.; Shenglia, N.; Gigineishvili, T.; Gigineishvili, D. Geographical variation in malignant and benign/borderline brain and CNS tumor incidence: A comparison between a high-income and a middle-income country. J. Neurooncol. 2020, 149, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.; Ugiliweneza, B.; Woo, S.; Skirboll, S.; Boaky, M. A Surveillance, Epidemiology and End Results-Medicare data analysis of elderly patients with glioblastoma multiforme: Treatment patterns, outcomes and cost. Mol. Clin. Oncol. 2015, 3, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, L.T.; Guo, H.R.; Chang, Y.K.; Lu, N.M.; Ho, S.Y. Clinical implications of multiple glioblastomas: An analysis of prognostic factors and survival to distinguish from their single counterparts. J. Formos. Med. Assoc. 2020, 119, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Cheo, S.T.T.; Lim, G.H.; Lim, K.H.C. Glioblastoma multiforme outcomes of 107 patients treated in two Singapore institutions. Singap. Med. J. 2017, 58, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Narita, Y.; Shibui, S. Trends and outcomes in the treatment of gliomas based on data during 2001–2004 from the brain tumor registry of Japan. Neurol. Med. Chir. 2015, 55, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Shi, Q.; Li, M.; Nagamuthu, C.; Andres, E.; Davis, F.G. Canadian brain cancer survival rates by tumour type and region: 1992–2008. Can. J. Public Health 2016, 107, e37–e42. [Google Scholar] [CrossRef] [Green Version]
- Cote, D.J.; Ostrom, Q.T.; Gittleman, H.; Duncan, K.R.; CreveCoeur, T.S.; Kruchko, C.; Smith, T.R.; Stampfer, M.J.; Barnholtz-Sloan, J.S. Glioma incidence and survival variations by county-level socioeconomic measures. Cancer 2019, 125, 3390–3400. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Weller, R.O.; Garfield, J.S. Epidemiology of primary tumours of the brain and spinal cord: A regional survey in southern England. J. Neurol. Neurosurg. Psychiatry 1976, 39, 290–296. [Google Scholar] [CrossRef]
- Walker, E.V.; Ross, J.; Yuan, Y.; Smith, T.R.; Davis, F.G. Brain cancer survival in Canada 1996–2008: Effects of sociodemographic characteristics. Curr. Oncol. 2019, 26, e292–e299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, D.S.; Gallo, V.; Schlehofer, B.; Tjønneland, A.; Olsen, A.; Overvad, K.; Dahm, C.C.; Kaaks, R.; Lukanova, A.; Boeing, H.; et al. Reproductive factors and exogenous hormone use in relation to risk of glioma and meningioma in a large European cohort study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2562–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigertz, A.; Lönn, S.; Mathiesen, T.; Ahlbom, A.; Hall, P.; Feychting, M. Risk of brain tumors associated with exposure to exogenous female sex hormones. Am. J. Epidemiol. 2006, 164, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, H.; Strandéus, M.; Milos, P.; Hallbeck, M.; Vrethem, M.; Lind, J. Improved survival of Swedish glioblastoma patients treated according to Stupp. Acta Neurol. Scand. 2018, 138, 332–337. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Ho, V.K.Y.; Zwinderman, A.H.; Ackermans, L.; Ardon, H.; Boomstra, S.; Bouwknegt, W.; van den Brink, W.A.; Dirven, C.M.; van der Gaag, N.A.; et al. Between-hospital variation in mortality and survival after glioblastoma surgery in the Dutch Quality Registry for Neuro Surgery. J. Neurooncol. 2019, 144, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Helseth, A.; Mork, S.J. Neoplasms of the central nervous system in Norway. III. Epidemiological characteristics of intracranial gliomas according to histology. Apmis 1989, 97, 547–555. [Google Scholar] [CrossRef]
- Nomura, E.; Ioka, A.; Tsukuma, H. Trends in the incidence of primary intracranial tumors in Osaka, Japan. Jpn. J. Clin. Oncol. 2011, 41, 291–294. [Google Scholar] [CrossRef]
- Wiedmann, M.K.H.; Brunborg, C.; Di Ieva, A.; Lindemann, K.; Johannesen, T.B.; Vatten, L.; Helseth, E.; Zwart, J.A. The impact of body mass index and height on the risk for glioblastoma and other glioma subgroups: A large prospective cohort study. Neuro-Oncology 2017, 19, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.C.; Yang, S.; Liu, X.Y.; Zhao, Y.X. Effect of marital status on survival in glioblastoma multiforme by demographics, education, economic factors, and insurance status. Cancer Med. 2018, 7, 3722–3742. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, P.; Meneghini, F.; Grigoletto, F.; Gerosa, M.; Licata, C.; Casentini, L.; Longatti, P.L.; Padoan, A.; Mingrino, S. Risk factors for cerebral glioma in adults: A case-control study in an Italian population. J. Neurooncol. 1994, 19, 61–67. [Google Scholar] [CrossRef]
- Guo, Y.; Su, Z.Y.; Zhang, C.; Gaspar, J.M.; Wang, R.; Hart, R.P.; Verzi, M.P.; Kong, A.N.T. Mechanisms of colitis-accelerated colon carcinogenesis and its prevention with the combination of aspirin and curcumin: Transcriptomic analysis using RNA-seq. Biochem. Pharmacol. 2017, 135, 22–34. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Loh, J.K.; Hwang, S.L.; Lieu, A.S.; Huang, T.Y.; Howng, S.L. The alteration of prostaglandin E2 levels in patients with brain tumors before and after tumor removal. J. Neurooncol. 2002, 57, 147–150. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Elmaci, I.; Cengiz, S.; Emekli-Alturfan, E.; Ozpinar, A. From epidemiology to treatment: Aspirin’s prevention of brain and breast-cancer and cardioprotection may associate with its metabolite gentisic acid. Chem. Biol. Interact. 2018, 291, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Scheurer, M.E.; Amirian, E.S.; Davlin, S.L.; Rice, T.; Wrensch, M.; Bondy, M.L. Effects of antihistamine and anti-inflammatory medication use on risk of specific glioma histologies. Int. J. Cancer 2011, 129, 2290–2296. [Google Scholar] [CrossRef] [Green Version]
- Sivak-Sears, N.R.; Schwartzbaum, J.A.; Miike, R.; Moghadassi, M.; Wrensch, M. Case-control study of use of nonsteroidal antiinflammatory drugs and glioblastoma multiforme. Am. J. Epidemiol. 2004, 159, 1131–1139. [Google Scholar] [CrossRef]
- Ferris, J.S.; McCoy, L.; Neugut, A.I.; Wrensch, M.; Lai, R. HMG CoA reductase inhibitors, NSAIDs and risk of glioma. Int. J. Cancer 2012, 131, E1031–E1037. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, S.E.; Moore, S.C.; Pfeiffer, R.M.; Inskip, P.D.; Park, Y.; Hollenbeck, A.; Rajaraman, P. Nonsteroidal anti-inflammatory drugs and glioma in the NIH-AARP diet and health study cohort. Cancer Prev. Res. 2011, 4, 2027–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhns, R.P.; James, W.S.; Torabi, M.; Borgstrom, M.; Roussas, A.; Lemole, M. Survival as a Function of Nonsteroidal Anti-inflammatory Drug Use in Patients with Glioblastoma. Cureus 2018, 10, e3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheurer, M.E.; El-Zein, R.; Thompson, P.A.; Aldape, K.D.; Levin, V.A.; Gilbert, M.R.; Weinberg, J.S.; Bondy, M.L. Long-term anti-inflammatory and antihistamine medication use and adult glioma risk. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1277–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlehofer, B.; Blettner, M.; Preston-Martin, S.; Preston-Martin, S.; Niehoff, D.; Wahrendorf, J.; Arslan, A.; Ahlbom, A.; Choi, W.N.; Giles, G.G.; et al. Role of medical history in brain tumour development. Results from the international adult brain tumour study. Int. J. Cancer 1999, 82, 155–160. [Google Scholar] [CrossRef]
- Schoemaker, M.J.; Swerdlow, A.J.; Hepworth, S.J.; McKinney, P.A.; van Tongeren, M.; Muir, K.R. History of allergies and risk of glioma in adults. Int. J. Cancer 2006, 119, 2165–2172. [Google Scholar] [CrossRef]
- McCarthy, B.J.; Rankin, K.; Il’yasova, D.; Erdal, S.; Vick, N.; Ali-Osman, F.; Bigner, D.D.; Davis, F. Assessment of type of allergy and antihistamine use in the development of glioma. Cancer Epidemiol. Biomark. Prev. 2011, 20, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Lu, Q.; Lenahan, C.; Yang, S.; Zhou, D.; Qi, X. Whether statin use improves the survival of patients with glioblastoma?: A meta-analysis. Medicine 2020, 99, e18997. [Google Scholar] [CrossRef]
- Cote, D.J.; Rosner, B.A.; Smith-Warner, S.A.; Egan, K.M.; Stampfer, M.J. Statin use, hyperlipidemia, and risk of glioma. Eur. J. Epidemiol. 2019, 34, 997–1011. [Google Scholar] [CrossRef]
- Rendon, L.F.; Tewarie, I.A.; Cote, D.J.; Gabriel, A.; Smith, T.R.; Broekman, M.L.D.; Mekary, R.A. Statins and Gliomas: A Systematic Review of the Preclinical Studies and Meta-Analysis of the Clinical Literature. Drugs 2022, 82, 293–310. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.; Kaminska, B. Cannabinoid signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 209–220. [Google Scholar]
- Ramer, R.; Hinz, B. Cannabinoids as Anticancer Drugs. Adv. Pharmacol. 2017, 80, 397–436. [Google Scholar] [PubMed]
- Guzmán, M.; Duarte, M.J.; Blázquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.C.M.; Dos Santos Júnior, J.G.; Stefano, S.C.; Da Silveira, D.X. Systematic review of the literature on clinical and experimental trials on the antitumor effects of cannabinoids in gliomas. J. Neurooncol. 2014, 116, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [Green Version]
- Massi, P.; Valenti, M.; Solinas, M.; Parolaro, D. Molecular mechanisms involved in the antitumor activity of cannabinoids on gliomas: Role for oxidative stress. Cancers 2010, 2, 1013–1026. [Google Scholar] [CrossRef]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol enhances the inhibitory effects of Δ9- tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol. Cancer Ther. 2010, 9, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, S.; Rheinstein, P.H.; Rosenzweig, K.E. Allergy may confer better survival on patients with gliomas. Clin. Neurol. Neurosurg. 2019, 177, 63–67. [Google Scholar] [CrossRef]
- Schwartzbaum, J.; Ahlborn, A.; Malmer, B.; Lönn, S.; Brookes, A.J.; Doss, H.; Debinski, W.; Henriksson, R.; Feychting, M. Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res. 2005, 65, 6459–6465. [Google Scholar] [CrossRef] [Green Version]
- Linos, E.; Raine, T.; Alonso, A.; Michaud, D. Atopy and risk of brain tumors: A meta-analysis. J. Natl. Cancer Inst. 2007, 99, 1544–1550. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbaum, J.; Ding, B.; Johannesen, T.B.; Osnes, L.T.N.; Karavodin, L.; Ahlbom, A.; Feychting, M.; Grimsrud, T.K. Association between prediagnostic IgE levels and risk of glioma. J. Natl. Cancer Inst. 2012, 104, 1251–1259. [Google Scholar] [CrossRef]
- Blowers, L.; Preston-Martin, S.; Mack, W.J. Dietary and other lifestyle factors of women with brain gliomas in Los Angeles County (California, USA). Cancer Causes Control 1997, 8, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Cantor, K.P.; Zhang, Y.; Chiu, B.C.H.; Lynch, C.F. Risk of brain glioma not associated with cigarette smoking or use of other tobacco products in Iowa. Cancer Epidemiol. Biomark. Prev. 2001, 10, 413–414. [Google Scholar]
- Lachance, D.H.; Yang, P.; Johnson, D.R.; Decker, P.A.; Kollmeyer, T.M.; McCoy, L.S.; Rice, T.; Xiao, Y.; Ali-Osman, F.; Wang, F.; et al. Associations of high-grade glioma with glioma risk alleles and histories of allergy and smoking. Am. J. Epidemiol. 2011, 174, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Patel, B.; Patri, M. Adolescence benzo[a]pyrene treatment induces learning and memory impairment and anxiolytic like behavioral response altering neuronal morphology of hippocampus in adult male Wistar rats. Toxicol. Rep. 2019, 6, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sharma, S.; Zhu, L.X.; Kogai, T.; Hershman, J.M.; Brent, G.A.; Dubinett, S.M.; Huang, M. Nonradioactive iodide effectively induces apoptosis in genetically modified lung cancer cells. Cancer Res. 2003, 63, 5065–5072. [Google Scholar] [PubMed]
- Humans, IWGotEoCRt, and IARC Working Group. Tobacco smoke and involuntary smoking. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 83, 1–1438. [Google Scholar]
- Jakszyn, P.; Agudo, A.; Berenguer, A.; Ibáñez, R.; Amiano, P.; Pera, G.; Ardanaz, E.; Barricarte, A.; Chirlaque, M.D.; Dorronsoro, M.; et al. Intake and food sources of nitrites and N-nitrosodimethylamine in Spain. Public Health Nutr. 2006, 9, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricker, A.R.; Pfundstein, B.; Theobald, E.; Preussmann, R.; Spiegelhalder, B. Mean daily intake of volatile N-nitrosamines from foods and beverages in West Germany in 1989–1990. Food Chem. Toxicol. 1991, 29, 729–732. [Google Scholar] [CrossRef]
- Anderson, L.M.; Souliotis, V.L.; Chhabra, S.K.; Moskal, T.J.; Harbaugh, S.D.; Kyrtopoulos, S.A. N-nitrosodimethylamine-derived O(6)-methylguanine in DNA of monkey gastrointestinal and urogenital organs and enhancement by ethanol. Int. J. Cancer 1996, 66, 130–134. [Google Scholar] [CrossRef]
- Honikel, K.O. The use and control of nitrate and nitrite for the processing of meat products. Meat Sci. 2008, 78, 68–76. [Google Scholar] [CrossRef]
- Kaplan, S.; Novikov, I.; Modan, B. Nutritional factors in the etiology of brain tumors: Potential role of nitrosamines, fat, and cholesterol. Am. J. Epidemiol. 1997, 146, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, D.S.; Holick, C.N.; Batchelor, T.T.; Giovannucci, E.; Hunter, D.J. Prospective study of meat intake and dietary nitrates, nitrites, and nitrosamines and risk of adult glioma. Am. J. Clin. Nutr. 2009, 90, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Saneei, P.; Willett, W.; Esmaillzadeh, A. Red and processed meat consumption and risk of glioma in adults: A systematic review and meta-analysis of observational studies. J. Res. Med. Sci. 2015, 20, 602–612. [Google Scholar] [PubMed]
- Fukushima, T.; Favereaux, A.; Huang, H.; Shimizu, T.; Yonekawa, Y.; Nakazato, Y.; Ohagki, H. Genetic alterations in primary glioblastomas in Japan. J. Neuropathol. Exp. Neurol. 2006, 65, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabihkhani, M.; Telesca, D.; Movassaghi, M.; Naeini, Y.B.; Naeini, K.M.; Hojat, S.A.; Gupta, D.; Lucey, G.M.; Ontiveros, M.; Wang, M.W.; et al. Incidence, survival, pathology, and genetics of adult Latino Americans with glioblastoma. J. Neurooncol. 2017, 132, 351–358. [Google Scholar] [CrossRef]
- Ron, E.; Modan, B.; Boice, J.D.; Alfandary, E.; Stovall, M.; Chetrit, A.; Katz, L. Tumors of the Brain and Nervous System after Radiotherapy in Childhood. N. Engl. J. Med. 1988, 319, 1033–1039. [Google Scholar] [CrossRef]
- Bowers, D.C.; Nathan, P.C.; Constine, L.; Woodman, C.; Bhatia, S.; Keller, K.; Bashore, L. Subsequent neoplasms of the CNS among survivors of childhood cancer: A systematic review. Lancet Oncol. 2013, 14, e321–e328. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.S.; Salotti, J.A.; Little, M.P.; McHugh, K.; Lee, C.; Kim, K.P.; Howe, N.L.; Ronckers, C.M.; Rajaraman, P.; Craft, A.W.; et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet 2012, 380, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Simińska, D.; Kojder, K.; Jeżewski, D.; Kojder, I.; Skórka, M.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Pathophysiology of Post-Traumatic Glioma. Int. J. Mol. Sci. 2018, 19, 2445. [Google Scholar] [CrossRef] [Green Version]
- Inskip, P.D.; Mellemkjaer, L.; Gridley, G.; Olsen, J.H. Incidence of intracranial tumors following hospitalization for head injuries (Denmark). Cancer Causes Control 1998, 9, 109–116. [Google Scholar] [CrossRef]
- Preston-Martin, S.; Paganini-Hill, A.; Henderson, B.E.; Pike, M.C.; Wood, C. Case-control study of intracranial meningiomas in women in Los Angeles County, California. J. Natl. Cancer Inst. 1980, 65, 67–73. [Google Scholar] [PubMed]
- Preston-Martin, S.; Pogoda, J.M.; Schlehofer, B.; Blettner, M.; Howe, G.R.; Ryan, P.; Menegoz, F.; Giles, G.G.; Rodvall, Y.; Choi, N.W.; et al. An international case-control study of adult glioma and meningioma: The role of head trauma. Int. J. Epidemiol. 1998, 27, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef]
- Jialal, I.; Devaraj, S. Subcutaneous adipose tissue biology in metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 2018, 33, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Little, R.B.; Madden, M.H.; Thompson, R.C.; Olson, J.J.; LaRocca, R.V.; Pan, E.; Browning, J.E.; Egan, K.M.; Nabors, L.B. Anthropometric factors in relation to risk of glioma. Cancer Causes Control 2013, 24, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919. [Google Scholar] [CrossRef]
- Moore, S.C.; Rajaraman, P.; Dubrow, R.; Darefsky, A.S.; Koebnick, C.; Hollenbeck, A.; Schatzkin, A.; Leitzmann, M.F. Height, body mass index, and physical activity in relation to glioma risk. Cancer Res. 2009, 69, 8349–8355. [Google Scholar] [CrossRef] [Green Version]
- Seliger, C.; Ricci, C.; Meier, C.R.; Bodmer, M.; Jick, S.S.; Bogdahn, U.; Hau, P.; Leitzmann, M.F. Diabetes, use of antidiabetic drugs, and the risk of glioma. Neuro-Oncology 2016, 18, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Lv, G.; Chen, W.; Jiang, J.; Wang, J. Height and kidney cancer risk: A meta-analysis of prospective studies. J. Cancer Res. Clin. Oncol. 2015, 141, 1799–1807. [Google Scholar] [CrossRef]
- Song, X.; Gong, X.; Zhang, T.; Jiang, W. Height and risk of colorectal cancer: A meta-analysis. Eur. J. Cancer Prev. 2018, 27, 521–529. [Google Scholar] [CrossRef]
- Juul, A.; Bang, P.; Hertel, N.T.; Main, K.; Dalgaard, P.; Jørgensen, K.; Müller, J.; Hall, K.; Skakkebaek, N.E. Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: Relation to age, sex, stage of puberty, testicular size, and body mass index. J. Clin. Endocrinol. Metab. 1994, 78, 744–752. [Google Scholar] [PubMed]
- Patil, S.S.; Railkar, R.; Swain, M.; Atreya, H.S.; Dighe, R.R.; Kondaiah, P. Novel anti IGFBP2 single chain variable fragment inhibits glioma cell migration and invasion. J. Neurooncol. 2015, 123, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Phillips, L.M.; Zhou, X.; Cogdell, D.E.; Chua, C.Y.; Huisinga, A.; Hess, K.R.; Fuller, G.N.; Zhang, W. Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J. Pathol. 2016, 239, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, S.M.; Celestino, J.; Wang, H.; Jiang, R.; Holland, E.C.; Fuller, G.N.; Zhang, W. Insulin-like growth factor binding protein 2 promotes glioma development and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Sunderman, F.W. Nasal toxicity, carcinogenicity, and olfactory uptake of metals. Ann. Clin. Lab. Sci. 2001, 31, 3–24. [Google Scholar]
- Parent, M.E.; Turner, M.C.; Lavoué, J.; Richard, H.; Figuerola, J.; Kincl, L.; Richardson, L.; Benke, G.; Blettner, M.; Fleming, S.; et al. Lifetime occupational exposure to metals and welding fumes, and risk of glioma: A 7-country population-based case-control study. Environ. Health Glob. Access Sci. Source 2017, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic effects and biomarkers of lead exposure: A review. Rev. Environ. Health 2009, 24, 15–45. [Google Scholar] [CrossRef]
- Liao, L.M.; Friesen, M.C.; Xiang, Y.B.; Cai, H.; Koh, D.H.; Ji, B.T.; Yang, G.; Li, H.L.; Locke, S.J.; Rothman, N.; et al. Occupational lead exposure and associations with selected cancers: The Shanghai men’s and women’s health study cohorts. Environ. Health Perspect. 2016, 124, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Caffo, M.; Caruso, G.; La Fata, G.; Barresi, V.; Visalli, M.; Venza, M.; Venza, I. Heavy Metals and Epigenetic Alterations in Brain Tumors. Curr. Genom. 2015, 15, 457–463. [Google Scholar] [CrossRef]
- Bhatti, P.; Stewart, P.A.; Hutchinson, A.; Rothman, N.; Linet, M.S.; Inskip, P.D.; Rajaraman, P. Lead exposure, polymorphisms in genes related to oxidative stress, and risk of adult brain tumors. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1841–1848. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Park, M.Y.; Kang, M.Y.; Shin, I.S.; An, S.; Kim, H.R. Occupational lead exposure and brain tumors: Systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2020, 17, 3975. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, P.; Stewart, P.A.; Samet, J.M.; Schwartz, B.S.; Linet, M.S.; Zahm, S.H.; Rothman, N.; Yeager, M.; Fine, H.A.; Black, P.M.; et al. Lead, genetic susceptibility, and risk of adult brain tumors. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, R.L.; Foran, C.M. Influence of persistent contaminants and steroid hormones on glioblastoma cell growth. J. Toxicol. Environ. Health-Part A Curr. Issues 2007, 70, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.I.; Semenciw, R.M.; Morison, D.; Magwood, S.; Mao, Y. Brain cancer and farming in Western Canada. Neuroepidemiology 1992, 11, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Doğanlar, O.; Doğanlar, Z.B.; Kurtdere, A.K.; Chasan, T.; Ok, E.S. Chronic exposure of human glioblastoma tumors to low concentrations of a pesticide mixture induced multidrug resistance against chemotherapy agents. Ecotoxicol. Environ. Saf. 2020, 202, 110940. [Google Scholar] [CrossRef]
- Kuan, A.S.; Green, J.; Kitahara, C.M.; De González, A.B.; Key, T.; Reeves, G.K.; Flou, S.; Balkwill, A.; Bradbury, K.; Liao, L.M.; et al. Diet and risk of glioma: Combined analysis of 3 large prospective studies in the UK and USA. Neuro-Oncology 2019, 21, 944–952. [Google Scholar] [CrossRef]
- Nkondjock, A. Coffee consumption and the risk of cancer: An overview. Cancer Lett. 2009, 277, 121–125. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Cavin, C.; Holzhaeuser, D.; Scharf, G.; Constable, A.; Huber, W.W.; Schilter, B. Cafestol and kahweol, two coffee specific diterpenes with anticarcinogenic activity. Food Chem. Toxicol. 2002, 40, 1155–1163. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea Polyphenol (-)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates Methylation-Silenced Genes in Cancer Cell Lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Huber, W.W.; Scharf, G.; Nagel, G.; Prustomersky, S.; Schulte-Hermann, R.; Kaina, B. Coffee and its chemopreventive components Kahweol and Cafestol increase the activity of O6-methylguanine-DNA methyltransferase in rat liver-Comparison with phase II xenobiotic metabolism. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2003, 522, 57–68. [Google Scholar] [CrossRef]
- Holick, C.N.; Smith, S.G.; Giovannucci, E.; Michaud, D.S. Coffee, tea, caffeine intake, and risk of adult glioma in three prospective cohort studies. Cancer Epidemiol. Biomark. Prev. 2010, 19, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrow, R.; Darefsky, A.S.; Freedman, N.D.; Hollenbeck, A.R.; Sinha, R. Coffee, tea, soda, and caffeine intake in relation to risk of adult glioma in the NIH-AARP Diet and Health Study. Cancer Causes Control 2012, 23, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creed, J.H.; Smith-Warner, S.A.; Gerke, T.A.; Egan, K.M. A prospective study of coffee and tea consumption and the risk of glioma in the UK Biobank. Eur. J. Cancer 2020, 129, 123–131. [Google Scholar] [CrossRef]
- Cote, D.J.; Bever, A.M.; Wilson, K.M.; Smith, T.R.; Smith-Warner, S.A.; Stampfer, M.J. A prospective study of tea and coffee intake and risk of glioma. Int. J. Cancer 2020, 146, 2442–2449. [Google Scholar] [CrossRef]
- Michaud, D.S.; Gallo, V.; Schlehofer, B.; Tjønneland, A.; Olsen, A.; Overvad, K.; Dahm, C.C.; Teucher, B.; Lukanova, A.; Boeing, H.; et al. Coffee and tea intake and risk of brain tumors in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am. J. Clin. Nutr. 2010, 92, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Malerba, S.; Galeone, C.; Pelucchi, C.; Turati, F.; Hashibe, M.; La Vecchia, C.; Tavani, A. A meta-analysis of coffee and tea consumption and the risk of glioma in adults. Cancer Causes Control 2013, 24, 267–276. [Google Scholar] [CrossRef]
- Galeone, C.; Malerba, S.; Rota, M.; Bagnardi, V.; Negri, E.; Scotti, L.; Bellocco, R.; Corrao, G.; Boffetta, P.; La Vecchia, C.; et al. A meta-analysis of alcohol consumption and the risk of brain tumours. Ann. Oncol. 2013, 24, 514–523. [Google Scholar] [CrossRef]
- Baglietto, L.; Giles, G.G.; English, D.R.; Karahalios, A.; Hopper, J.L.; Severi, G. Alcohol consumption and risk of glioblastoma; evidence from the Melbourne collaborative cohort study. Int. J. Cancer 2011, 128, 1929–1934. [Google Scholar] [CrossRef]
- And, A.M.; Mitsumori, K. Spontaneous Occurrence and Chemical Induction of Neurogenic Tumors in Rats—Influence of Host Factors and Specificity of Chemical Structure. Crit. Rev. Toxicol. 1990, 20, 287–310. [Google Scholar] [CrossRef] [PubMed]
- Hurley, S.F.; McNeil, J.J.; Donnan, G.A.; Forbes, A.; Salzberg, M.; Giles, G.G. Tobacco smoking and alcohol consumption as risk factors for glioma: A case-control study in Melbourne, Australia. J. Epidemiol. Community Health 1996, 50, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.-Y.; Shao, C.; Yang, C.; Wang, Z.; Hui, G.-Z. Alcohol consumption and risk of glioma: A meta-analysis of 19 observational studies. Nutrients 2014, 6, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, D.J.; Samanic, C.M.; Smith, T.R.; Wang, M.; Smith-Warner, S.A.; Stampfer, M.J.; Egan, K.M. Alcohol intake and risk of glioma: Results from three prospective cohort studies. Eur. J. Epidemiol. 2021, 36, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Samanic, C.M.; Cote, D.J.; Creed, J.H.; Stampfer, M.J.; Wang, M.; Smith-Warner, S.A.; Egan, K.M. Prospective study of sleep duration and glioma risk. Cancer Causes Control. 2021, 32, 1039–1042. [Google Scholar] [CrossRef]
- Orešković, D.; Kaštelančić, A.; Raguž, M.; Dlaka, D.; Predrijevac, N.; Matec, D.; Matec, M.; Tomac, D.; Jeleč, V.; Marinović, T.; et al. The vicious interplay between disrupted sleep and malignant brain tumors: A narrative review. Croat. Med. J. 2021, 62, 376–386. [Google Scholar] [CrossRef]
- Lissoni, P.; Meregalli, S.; Nosetto, L.; Barni, S.; Tancini, G.; Fossati, V.; Maestroni, G. Increased survival time in brain glioblastomas by a radioneuroendocrine strategy with radiotherapy plus melatonin compared to radiotherapy alone. Oncology 1996, 53, 43–46. [Google Scholar] [CrossRef]
- Cutando, A.; López-Valverde, A.; Arias-Santiago, S.; De Vicente, J.; De Diego, R.G. Role of melatonin in cancer treatment. Anticancer Res. 2012, 32, 2747–2753. [Google Scholar]
- Martín, V.; García-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Sanchez-Sanchez, A.; Antolín, I.; Rodriguez, C. Melatonin sensitizes human malignant glioma cells against TRAIL-induced cell death. Cancer Lett. 2010, 287, 216–223. [Google Scholar] [CrossRef]
- Zheng, X.; Pang, B.; Gu, G.; Gao, T.; Zhang, R.; Pang, Q.; Liu, Q. Melatonin inhibits glioblastoma stem-like cells through suppression of EZH2-NOTCH1 signaling axis. Int. J. Biol. Sci. 2017, 132, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Ortner, D.; Tripp, C.H.; Komenda, K.; Dubrac, S.; Zelger, B.; Hermann, M.; Doppler, W.; Tymoszuk, P.Z.; Boon, L.; Clausen, B.E.; et al. Langerhans cells and NK cells cooperate in the inhibition of chemical skin carcinogenesis. Oncoimmunology 2016, 6, e1260215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Murata, M. Inflammation and cancer. Environ Health Prev. Med. 2018, 23, 50. [Google Scholar] [CrossRef] [Green Version]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.H.; Chen, Z.; Chen, K.; Liao, F.T.; Chung, C.E.; Liu, X.; Lin, Y.C.; Keohavong, P.; Leikauf, G.D.; Di, Y.P. Lipopolysaccharide-Mediated Chronic Inflammation Promotes Tobacco Carcinogen-Induced Lung Cancer and Determines the Efficacy of Immunotherapy. Cancer Res. 2021, 81, 144–157. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [Green Version]
- Kore, R.A.; Abraham, E.C. Inflammatory cytokines, interleukin-1 beta and tumor necrosis factor-alpha, upregulated in glioblastoma multiforme, raise the levels of CRYAB in exosomes secreted by U373 glioma cells. Biochem. Biophys. Res. Commun. 2014, 453, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Domon-Dell, C.; Kang, J.; Chung, D.H.; Freund, J.-N.; Evers, B.M. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappaB pathway is linked to a default IkappaB-alpha autoregulatory loop. J. Biol. Chem. 2004, 279, 4285–4291. [Google Scholar] [CrossRef] [Green Version]
- Teo, G.S.L.; Ankrum, J.A.; Martinelli, R.; Boetto, S.E.; Simms, K.; Sciuto, T.E.; Dvorak, A.M.; Karp, J.M.; Carman, C.V. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-α-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells 2012, 30, 2472–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Gong, K.; Ali, S.; Ali, N.; Shallwani, S.; Hatanpaa, K.J.; Pan, E.; Mickey, B.; Burma, S.; Wang, D.H.; et al. A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat. Neurosci. 2017, 20, 1074–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabors, L.B.; Suswam, E.; Huang, Y.; Yang, X.; Johnson, M.J.; King, P.H. Tumor Necrosis Factor α Induces Angiogenic Factor Up-Regulation in Malignant Glioma Cells: A Role for RNA Stabilization and HuR. Cancer Res. 2003, 63, 4181–4187. [Google Scholar]
- Tarassishin, L.; Casper, D.; Lee, S.C. Aberrant expression of interleukin-1β and inflammasome activation in human malignant gliomas. PLoS ONE 2014, 9, e103432. [Google Scholar]
- Tarassishin, L.; Lim, J.; Weatherly, D.B.; Angeletti, R.H.; Lee, S.C. Interleukin-1-induced changes in the glioblastoma secretome suggest its role in tumor progression. J. Proteom. 2014, 99, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Kharbanda, S.; Chen, R.; Soriano, R.H.; Aldape, K.; Misra, A.; Zha, J.; Forrest, W.F.; Nigro, J.M.; Modrusan, Z.; et al. Identification of IGF2 signaling through phosphoinositide-3-kinase regulatory subunit 3 as a growth-promoting axis in glioblastoma. Proc. Natl. Acad. Sci. USA 2007, 104, 3466–3471. [Google Scholar] [CrossRef] [Green Version]
- Tili, E.; Michaille, J.-J.; Wernicke, D.; Alder, H.; Costinean, S.; Volinia, S.; Croce, C.M. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4908–4913. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.W.; Binte Hanafi, Z.; Chew, L.C.Y.; Mei, Y.; Liu, H. IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells 2021, 10, 92. [Google Scholar] [CrossRef]
- Melisi, D.; Niu, J.; Chang, Z.; Xia, Q.; Peng, B.; Ishiyama, S.; Evans, D.B.; Chiao, P.J. Secreted interleukin-1alpha induces a metastatic phenotype in pancreatic cancer by sustaining a constitutive activation of nuclear factor-kappaB. Mol. Cancer Res. 2009, 7, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Tian, L.; Han, Y.; Vogelbaum, M.; Stark, G.R. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin-1 signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 4365–4370. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A.; Tamura, M.; Hasegawa, M.; Ishiuchi, S.; Hirato, J.; Nakazato, Y. Expression of interleukin-1beta mRNA and protein in human gliomas assessed by RT-PCR and immunohistochemistry. J. Neuropathol. Exp. Neurol. 1998, 57, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, B.D.; Moynagh, P.N. Persistent interleukin-1beta signaling causes long term activation of NFkappaB in a promoter-specific manner in human glial cells. J. Biol. Chem. 2006, 281, 10316–10326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meini, A.; Sticozzi, C.; Massai, L.; Palmi, M. A nitric oxide/Ca2+/calmodulin/ERK1/2 mitogen-activated protein kinase pathway is involved in the mitogenic effect of IL-1beta in human astrocytoma cells. Br. J. Pharmacol. 2008, 153, 1706–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paugh, B.S.; Bryan, L.; Paugh, S.W.; Wilczynska, K.M.; Alvarez, S.M.; Singh, S.K.; Kapitonov, D.; Rokita, H.; Wright, S.; Griswold-Prenner, I.; et al. Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J. Biol. Chem. 2009, 284, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Dixit, D.; Ghosh, S.; Sen, E. COX-2 regulates the proliferation of glioma stem like cells. Neurochem. Int. 2011, 59, 567–571. [Google Scholar] [CrossRef]
- Yeung, Y.T.; Bryce, N.S.; Adams, S.; Braidy, N.; Konayagi, M.; McDonald, K.L.; Teo, C.; Guillemin, G.J.; Grewal, T.; Munoz, L. p38 MAPK inhibitors attenuate pro-inflammatory cytokine production and the invasiveness of human U251 glioblastoma cells. J. Neurooncol. 2012, 109, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; He, X.; Song, W.; Han, D.; Niu, J.; Wang, J. Role of IL-6 in the invasiveness and prognosis of glioma. Int. J. Clin. Exp. Med. 2015, 8, 9114–9120. [Google Scholar]
- Tchirkov, A.; Khalil, T.; Chautard, E.; Mokhtari, K.; Véronèse, L.; Irthum, B.; Vago, P.; Kémény, J.-L.; Verrelle, P. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br. J. Cancer 2007, 96, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Crespo, S.; Kind, M.; Arcaro, A. The role of the PI3K/AKT/mTOR pathway in brain tumor metastasis. JCMT 2016, 2, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The role of JAK-STAT signaling within the CNS. JAKSTAT 2013, 2, e22925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baan, R.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Islami, F.; Galichet, L.; Straif, K.; et al. Carcinogenicity of radio frequency electromagnetic fields. Lancet Oncol. 2011, 12, 624–626. [Google Scholar] [CrossRef]
- Hardell, L.; Carlberg, M. Mobile phone and cordless phone use and the risk for glioma—Analysis of pooled case-control studies in Sweden, 1997–2003 and 2007–2009. Pathophysiology 2015, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, P.J.; Momoli, F.; Parent, M.É.; Siemiatycki, J.; Turner, M.C.; Krewski, D. Cell phone use and the risk of glioma: Are case-control study findings consistent with Canadian time trends in cancer incidence? Environ. Res. 2021, 200, 111283. [Google Scholar] [CrossRef] [PubMed]
- Cardis, E.; Deltour, I.; Vrijheid, M.; Combalot, E.; Moissonnier, M.; Tardy, H.; Armstrong, B.; Giles, G.; Brown, J.; Siemiatycki, J.; et al. Brain tumour risk in relation to mobile telephone use: Results of the INTERPHONE international case-control study. Int. J. Epidemiol. 2010, 39, 675–694. [Google Scholar]
- Olsson, A.; Bouaoun, L.; Auvinen, A.; Feychting, M.; Johansen, C.; Mathiesen, T.; Melin, B.; Lahkola, A.; Larjavaara, S.; Villegier, A.S.; et al. Survival of glioma patients in relation to mobile phone use in Denmark, Finland and Sweden. J. Neurooncol. 2019, 141, 139–149. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 93–101. [Google Scholar] [CrossRef]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current Trends in Targeted Therapies for Glioblastoma Multiforme. Neurol. Res. Int. 2012, 2012, 878425. [Google Scholar] [CrossRef]
- Roh, T.H.; Kang, S.G.; Moon, J.H.; Sung, K.S.; Park, H.H.; Kim, S.H.; Kim, E.H.; Hong, C.K.; Suh, C.O.; Chang, J.H. Survival benefit of lobectomy over gross-total resection without lobectomy in cases of glioblastoma in the noneloquent area: A retrospective study. J. Neurosurg. 2020, 132, 895–901. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Cao, Y.; Li, P.; Liang, B.; Sun, J.; Feng, E. Risk of subsequent cancer among pediatric, adult and elderly patients following a primary diagnosis of glioblastoma multiforme: A population-based study of the SEER database. Int. J. Neurosci. 2017, 127, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, P.; Lei, T.; Qu, L.; Huang, H.; Mu, Q. Acute lymphoblastic leukemia following temozolomide treatment in a patient with glioblastoma: A case report and review of the literature. Oncol. Lett. 2018, 15, 8663–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]





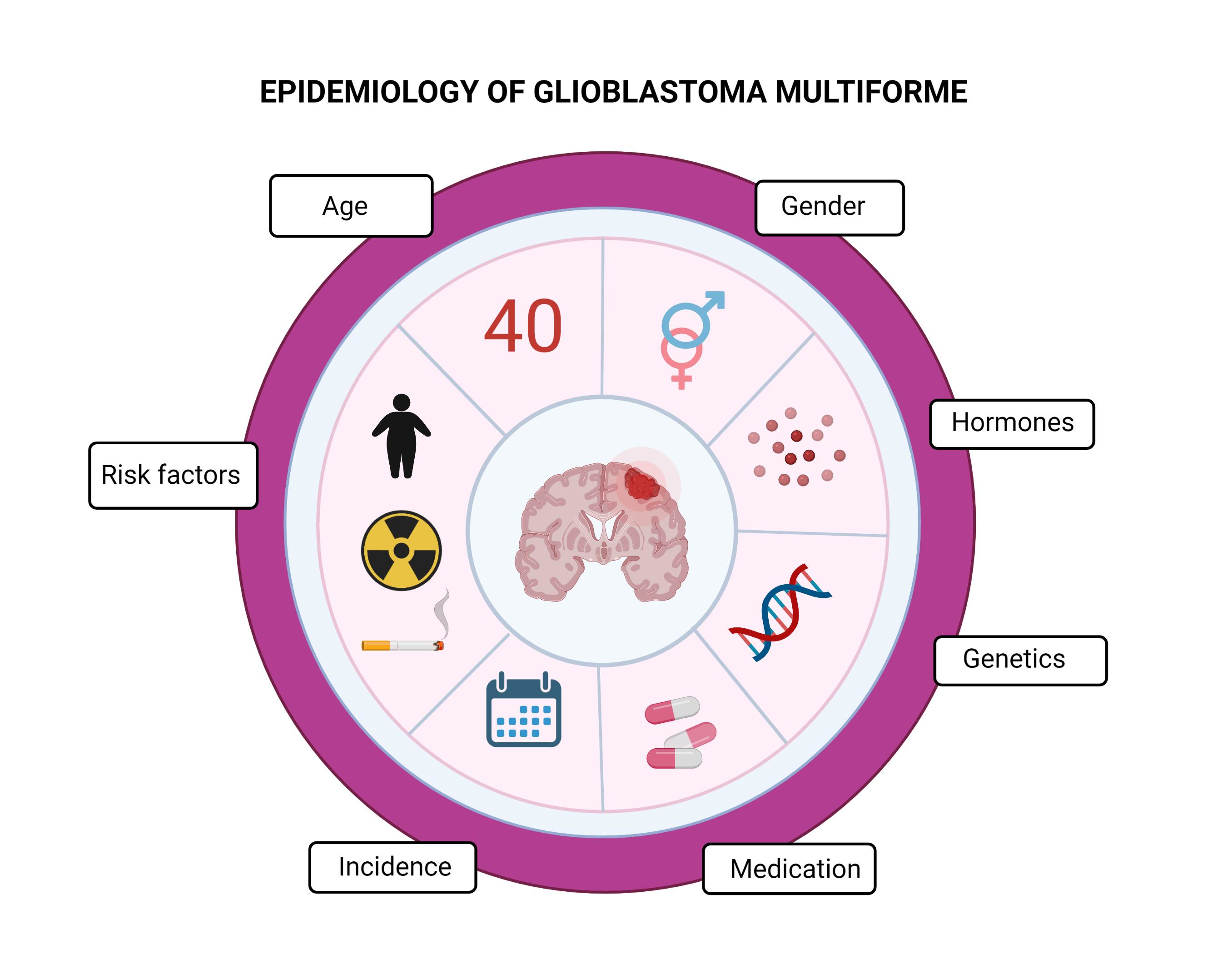{kind=link}
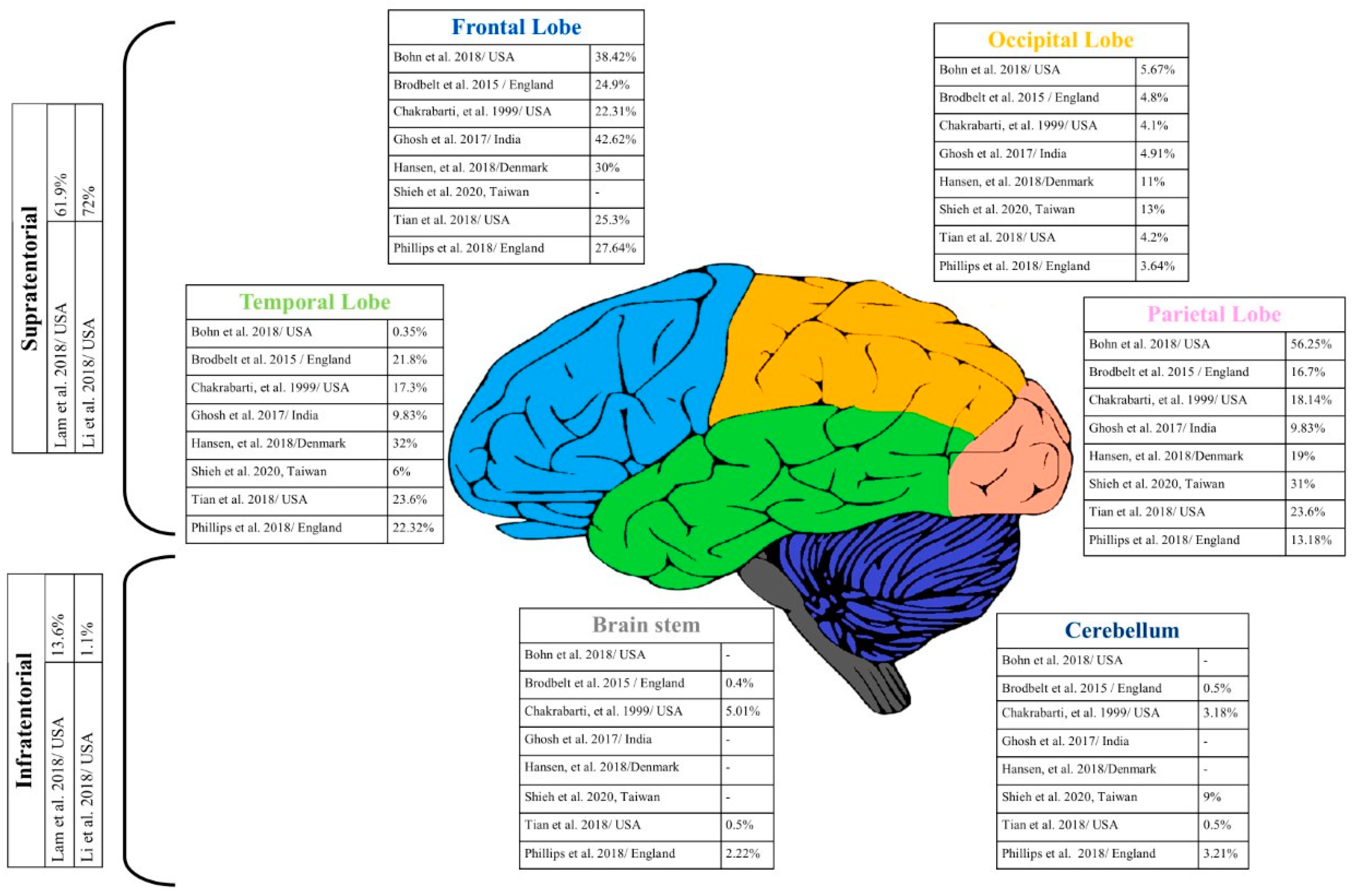{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Year Range | Country | Population Size (n) | Incidence/ 100,000/Year Male | Incidence/ 100,000/Year Female | Incidence/ 100,000/Year Male and Female | Age |
|---|---|---|---|---|---|---|---|
| Brodbelt et al., 2015 [29] | 2007–2011 | England | 10,743 | 5.87 (5.73–6.02) b | 3.54 (3.44–3.65) b | 4.64 (4.56–4.73) b | all |
| Chakrabarti et al., 2005 [30] | 1974–1999 | USA | 3832 | 2.68 (95% CI 2.56–2.80) a | 1.67 (95% CI 1.59–1.75) a | 2.11 (95% CI 2.0–2.17) a | ≥20 |
| Dho et al., 2017 [86] | 2013 | Republic of Korea | 19 | 0.12 j | 0.17 j | 0.87 j | 0–19 |
| 629 | 0.99 j | 0.78 j | all | ||||
| Dobec-Meić et al., 2006 [87] | 1996–2004 | Croatia | 63 | 5.1 g | 4.6 g | 4.8, (95% CI 3.7–6.2) g | ≥18 |
| Dobes et al., 2011 [88] | 2000–2008 | Australia | 2197 | - | - | 3.96 (3.37–4.52) a | all/not specified |
| Fabbro-Peray et al., 2019 [6] | 2008–2015 | France | 2053 | - | - | 3.3 f | all |
| Fleury et al., 1997 [89] | 1983–1990 | France | 764 | 3.09 b | 1.94 b | 2.38 b | all |
| Fuentes-Raspall et al., 2011 [90] | 1994–2005 | Spain | 134 | - | - | 1.59 d | all |
| Fuentes-Raspall et al., 2017 [91] | 1994–2013 | Spain | 463 | 5.05 (4.45; 5.72) e | 3.44 (2.97; 3.96) e | 4.17 (95% CI 3.80; 4.57) e | all |
| Gittleman et al., 2018 [92] | 2000–2014 | USA | 150,399 | 4.40 (4.38–4.42 95% CI) a | >20 | ||
| Gousias et al., 2009 [93] | 2005–2007 | Northwest Greece | 36 | 4.12 k | 3.26 k | 3.69 k | all |
| Hansen et al., 2018 [31] | 2009–2014 | Denmark | 1364 | 6.3 h | 3.9 h | - | 19–89 |
| Ho et al., 2014 [94] | 1989–2010 | Netherlands | 9504 | 3.2 b | 1.9 b | 2.5 b | ≥18 |
| Jazayeri et al., 2013 [95] | 2000–2009 | Iran | 3101 | - | - | 0.76 (0.70–0.83) g | all |
| Jung et al., 2013 [96] | 2010 | Republic of Korea | 523 | 0.89 j | 0.95 j | 0.77 j | all |
| Li et al., 2018 [32] | 1973–2014 | USA | 28,835 | - | - | 4.1 l | >20 |
| Larjavaara et al., 2007 [97] | 2000–2002 | Finland | 154 | - | - | 2 d | 20–69 |
| Natukka et al., 2019 [98] | 1990–2006 | Finland | 2284 | - | - | 3.8 (95% CI 3.7–4.0) e | all |
| Natukka et al., 2019 [98] | 2007–2016 | Finland | 1776 | - | - | 3.5 e | all |
| Ohgaki et al., 2004 [99] | 1980–1994 | Switzerland | 715 | 3.32 (CI, 2.69–4.09) d | 2.24 (CI, 1.56–3.22) d | - | all |
| Ostrom et al., 2013 [100] | 2006–2010 | USA | 50,872 | 3.97 a | 2.53 a | 3.19 a | all |
| Ostrom et al., 2017 [101] | 2010–2014 | USA | 56,421 | 3.99 (3.95–4.03 95% CI) a | 2.52 (2.49–2.56 95% CI) a | 3.20 (3.17–3.23 95% CI) a | all |
| Ostrom et al., 2020 [14] | 2013–2017 | USA | 60,056 | 4.03 (95% CI 3.98–4.07) a | 2.54 (95% CI 2.50–2.57) a | 3.23 (CI (95% 3.20–3.25) a | all |
| Schoenberg et al., 1976 [102] | 1935–1964 | Connecticut | 1167 | 2.07 i | 1.51 i | - | all |
| Walker et al., 2019 [103] | 2009–2013 | Canada | 5830 | 4.91 (95% CI 4.75–5.08) c | 3.24 (3.11; 3.37) c | 4.06 (3.95; 4.16) c | all |
| Wanner et al., 2020 [104] | 2009–2012 | Georgia | 72 | 0.59 (0.42; 0.82) a | 0.42 (0.29; 0.59) a | - | all |
| Reference | Year Range | Country | Population Size (n) | Survival Me (Months) | Survival (%). | |||
|---|---|---|---|---|---|---|---|---|
| 1 Year | 2 Years | 5 Years | 10 Years | |||||
| Brodbelt et al., 2015 [29] | 2007–2011 | England | 10,743 | 6.1 | 28.4 | 11.5 | 3.4 | - |
| Cheo et al., 2017 [107] | 2002–2011 | China | 107 | 15.1 | - | 23.5 | - | - |
| Fabbro-Peray et al., 2019 [6] | 2008–2015 | France | 2053 | 11.2 | 47.1 | 20.1 | 4.5 | - |
| Fuentes-Raspall et al., 2017 [91] | 1994–2013 | Spain | 463 | - | 24.0 | - | 3.3 | - |
| Ghosh et al., 2017 [25] | 2012–2014 | India | 61 | 8 | 19.15 | 3.27 | ||
| Gittleman et al., 2018 [92] | 2000–2014 | USA | 33,469 | - | 39.5 | 16.9 | 5.4 | 2.7 |
| Hansen et al., 2018 [31] | 2009–2014 | Denmark | 1364 | 11.2 | - | - | - | - |
| Lam et al., 2018 [24] | 2000–2010 | USA | 302 | 20 | - | 46.9 | - | - |
| Narita and Shibui, 2015 [108] | 2001–2004 | Japan | 1489 | 15 | - | - | 10.1 | - |
| Ostrom et al., 2017 [101] | 2000–2014 | USA | 33,951 | - | 39.7 | 17.2 | 5.5 | - |
| Yan Yuan et al., 2016 [109] | 1992–2008 | Canada | 14,120 | - | 26.5 | 9.5 | 4.0 | - |
| Reference | Year Range | Country | Population Size (n) | M:F Ratio |
|---|---|---|---|---|
| Bohn et al., 2018 [28] | 2010–2014 | USA | 3473 | 1.40 |
| Brodbelt et al., 2015 [29] | 2007–2011 | England | 10,743 | 1.66 |
| Bruhn et al., 2018 [115] | 2001–2012 | Sweden | 143 | 1.6 |
| Burton et al., 2015 [105] | 1997–2009 | USA | 3759 | 1.15 |
| Chakrabarti et al., 2005 [30] | 1974–1999 | USA | 3832 | 1.6 |
| Cheo et al., 2017 [107] | 2002–2011 | China | 107 | 1.55 |
| De Witt Hamer et al., 2019 [116] | 2011–2014 | Netherlands | 2382 | 1.63 |
| Dobec-Meić et al., 2006 [87] | 1996–2004 | Croatia Varazdin County | 63 | 1.12 |
| Dobes et al., 2011 [88] | 2000–2008 | Australia | 2197 | 1.6 |
| Fabbro-Peray et al., 2019 [6] | 2008–2015 | France | 2053 | 1.5 |
| Ghosh et al., 2017 [25] | 2012–2014 | India | 61 | 2.59 |
| Gousias et al., 2009 [93] | 2005–2007 | Northwest Greece | 36 | 1.25 |
| Hansen et al., 2018 [31] | 2009–2014 | Denmark | 1364 | 1.6 |
| Helseth and Mork, 1989 [117] | 1955–1984 | Norway | 2813 | 1.48 |
| Jung et al., 2013 [96] | 2010 | Republic of Korea | 523 | 1.22 |
| Li et al., 2018 [32] | 1973–2014 | USA | 28,835 | 1.35 |
| Nomura et al., 2011 [118] | 1995–2004 | Osaka, Japan | 713 | 1.25 |
| Ostrom et al., 2017 [101] | 2010–2014 | USA | 56,421 | 1.36 |
| Ostrom et al., 2013 [100] | 2006–2010 | USA | 50,872 | 1.57 |
| Shieh et al., 2020 [106] | 2005–2016 | Taiwan | 48 | 1.18 |
| Tian et al., 2018 [85] | 2000–2008 | USA | 6586 | 1.60 |
| Wiedmann et al., 2017 [119] | 1963–1975 | Norway | 3102 | 1.33 |
| J.-C. Xie et al., 2018 [120] | 2004–2015 | USA | 30,767 | 1.38 |
| Zampieri et al., 1994 [121] | 1986–1988 | Italy | 72 | 1.25 |
| Reference | Year Range | Country | Population Size (n) | Age | Race/Ethnicity (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| White | Black | Hispanic | Asian | Unknown/Other | |||||
| Bohn et al., 2018 [28] | 2010–2014 | USA | 3473 | ≥18 | 83.21 | 5.90 | 5.53 | 5.36 | - |
| Burton et al., 2015 [105] | 1997–2009 | USA | 3759 | >65 | 93.11 | 3.14 | - | - | 3.75 |
| Cheo et al., 2017 [107] | 2002–2011 | China | 107 | 13–85 | - | - | - | Chinese 76.6 Malay 9.3 Indian 7.5 | 6.5 |
| Li et al., 2018 [32] | 1973–2014 | USA | 28 835 | >20 | 91.4 | 4.7 | - | - | 3.9 |
| Ostrom et al., 2013 [100] | 2006–2010 | USA | 50,872 | all | 91.78 | 5.62 | 6.77 | 2.22 | 0.38 |
| Ostrom et al., 2017 [101] | 2010–2014 | USA | 56,421 | all | 91.11 | 6.12 | 7.34 | 2.38 | 0.39 |
| Ostrom et al., 2020 [14] | 2013–2017 | USA | 60,056 | all | 91.47 | 6.21 | 7.87 | 1.87 | 0.45 |
| Xie et al., 2018 [120] | 2004–2015 | USA | 30,767 | all | 89.6 | 5.5 | - | - | 4.7 |
| Reference | Year Range | Country | Population Size (n) | Age | Race/Ethnicity (Incidence per 100,000) | ||||
| White | Black | Hispanic | Asian | Unknown/Other | |||||
| Chakrabarti et al., 2005 [30] | 1974–1999 | USA | 3832 | >20 | Latino 1.83 (1.65–2.01 95% CI) Non-Latino white 2.53 (2.44–2.62 95% CI) | 1.45 (1.27–1.62 95% CI) | - | - | - |
| Ostrom et al., 2013 [100] | 2006–2010 | USA | 50,872 | all | 3.45 (3.41–3.48 95% CI) | 1.67 (1.60–1.73 95% CI) | 2.45 (2.36–2.53 95% CI) | 1.67 (1.57–1.78 95% CI) | 1.48 (1.26–1.72 95% CI) |
| Ostrom et al., 2017 [101] | 2010–2014 | USA | 56,421 | all | 3.46 (3.43–3.49 95% CI) | 1.79 (1.73–1.85 95% CI) | 2.42 (2.35- 2.5095% CI) | 1.47 (1.26–1.69 95% CI) | 1.61 (1.52–1.70 95% CI) |
| Ostrom et al., 2020 [14] | 2013–2017 | USA | 60,056 | all | 3.51 (3.45–2.54 95% CI) | 1.77 (1.71–1.83 95% CI) | 2.46 (2.39–2.53 95% CI) | 1.18 (1.11–1.25 95% CI) | 1.49 (1.30–1.69 95% CI |
| Shabihkhani et al., 2017 [166] | 2001–2011 | USA | 21,184 | - | 5.1 (95% CI 5.0–5.3) | - | 3.4 (95% CI 3.3–3.5) | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. https://doi.org/10.3390/cancers14102412
Grochans S, Cybulska AM, Simińska D, Korbecki J, Kojder K, Chlubek D, Baranowska-Bosiacka I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers. 2022; 14(10):2412. https://doi.org/10.3390/cancers14102412
Chicago/Turabian StyleGrochans, Szymon, Anna Maria Cybulska, Donata Simińska, Jan Korbecki, Klaudyna Kojder, Dariusz Chlubek, and Irena Baranowska-Bosiacka. 2022. "Epidemiology of Glioblastoma Multiforme–Literature Review" Cancers 14, no. 10: 2412. https://doi.org/10.3390/cancers14102412