
Endocrine Therapy-Resistant Breast Cancer Cells Are More Sensitive to Ceramide Kinase Inhibition and Elevated Ceramide Levels Than Therapy-Sensitive Breast Cancer Cells

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
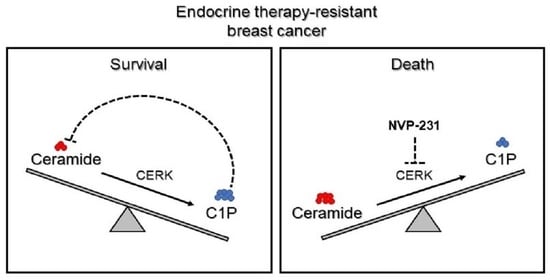
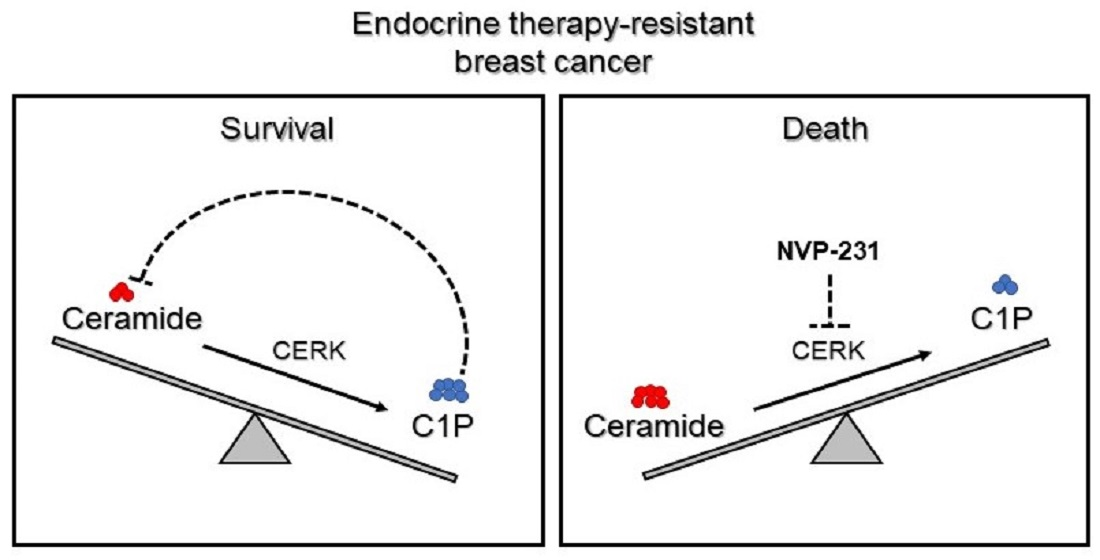{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Results
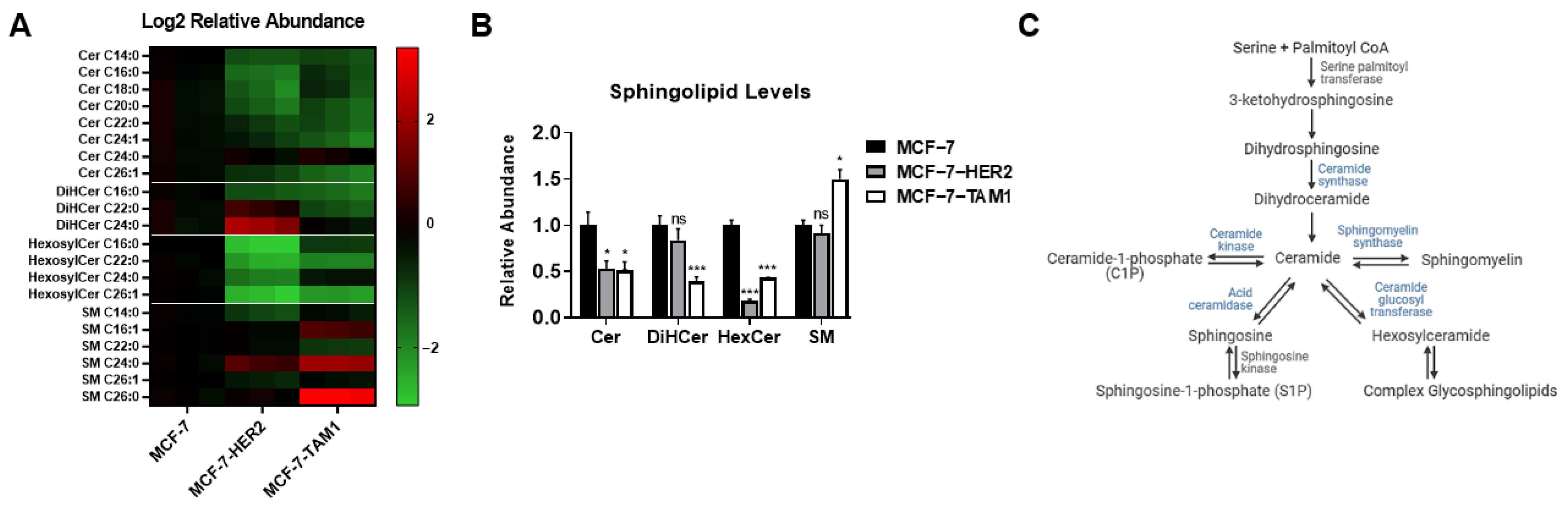
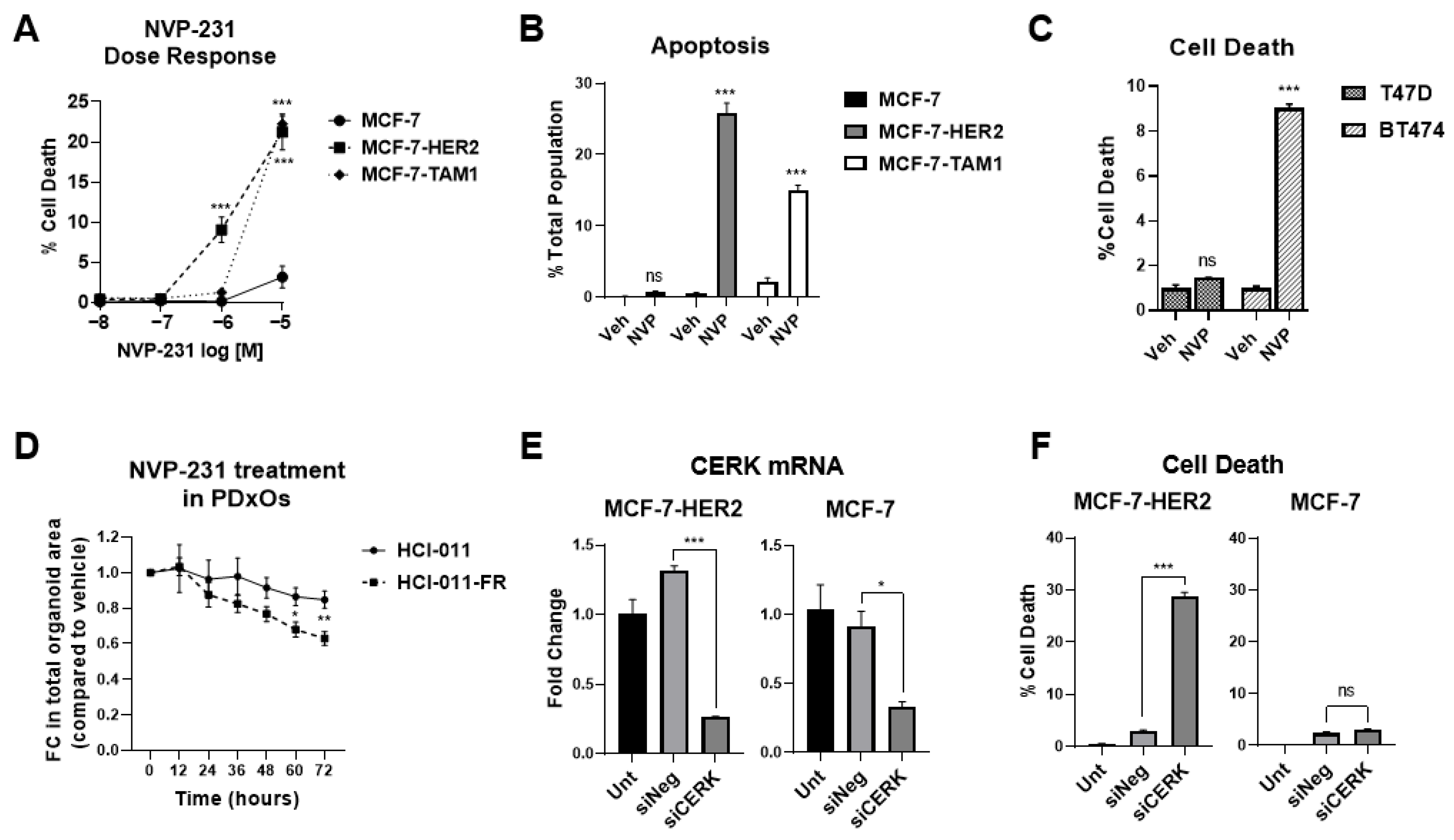
3.1.1. Tamoxifen-Resistant Cells Have an Altered Sphingolipidome and Rely on Ceramide Kinase (CERK) for Survival
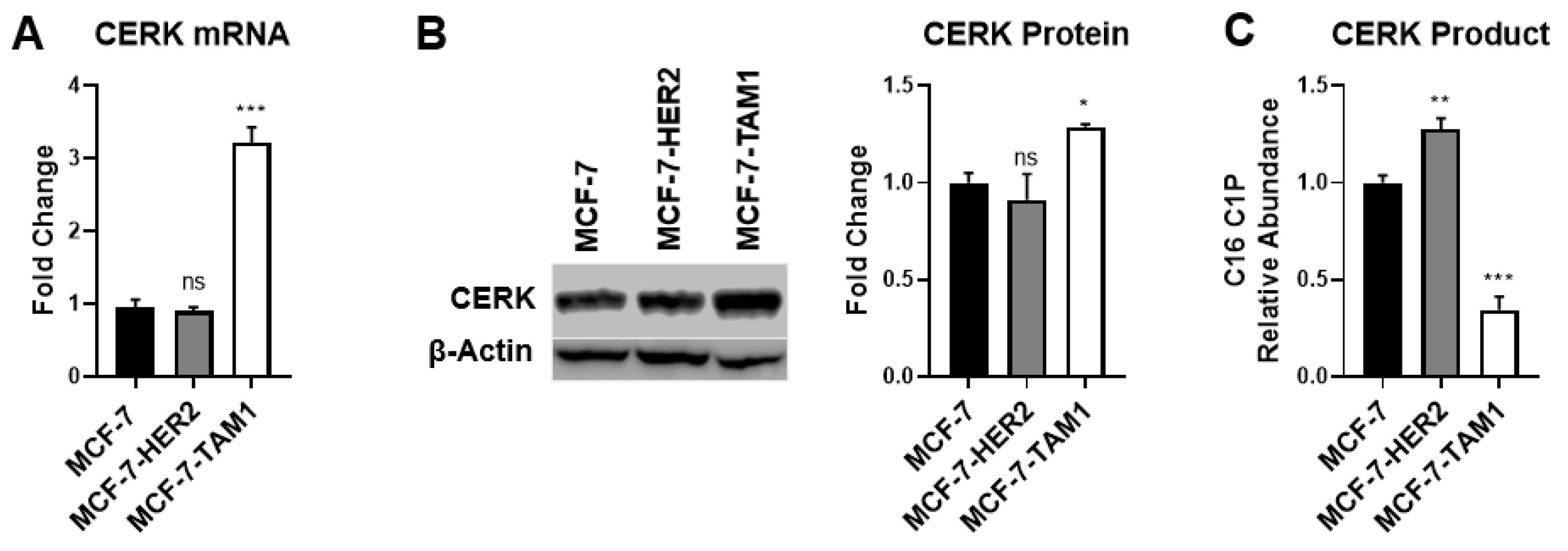
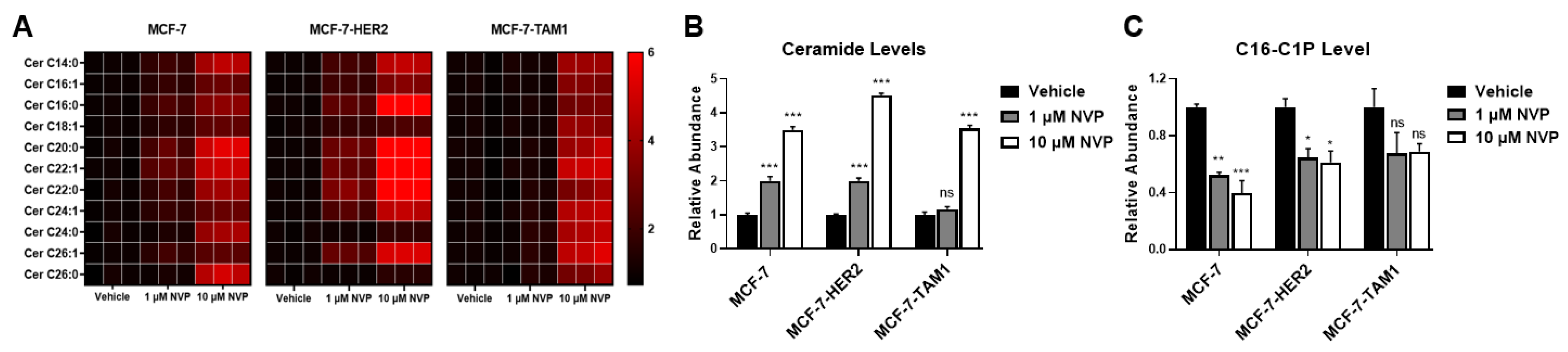
3.1.2. Differences in CERK Expression and Activity Does Not Explain Increased Sensitivity of Tamoxifen-Resistant Cells to CERK Inhibition
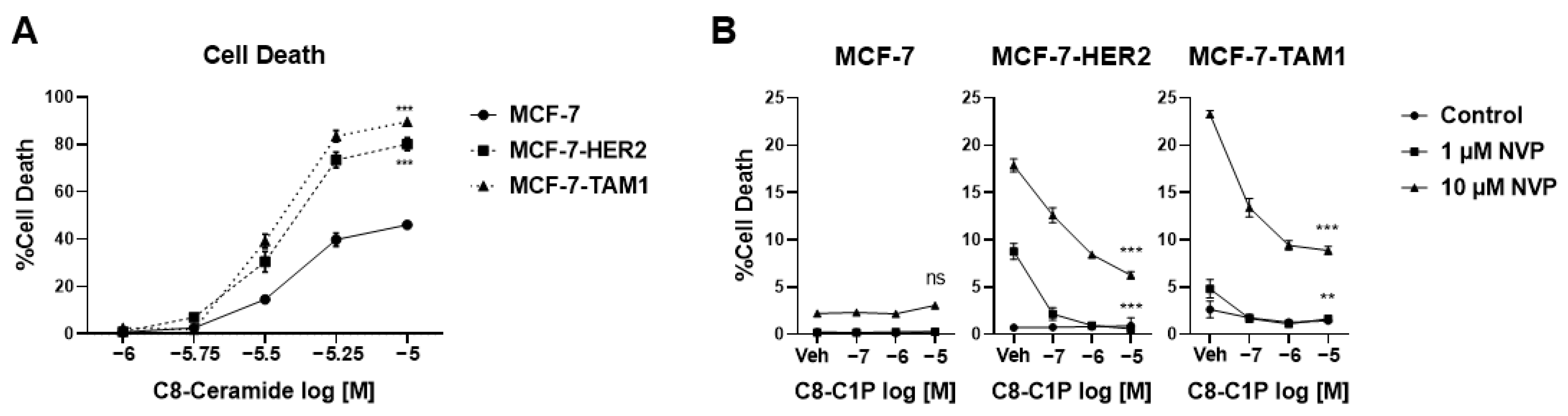
3.1.3. Tamoxifen-Resistant Cells Are More Sensitive to Ceramide-Induced Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- SEER Research Data in Released April 2021, Based on the November 2020 Submission; National Cancer Institute, DCCPS, Surveillance Research Program; National Cancer Institute: Bethesda, MD, USA, 2021.
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, I. Options in breast cancer local therapy: Who gets what? World J. Surg. 2012, 36, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, I.; Anderson, W.F.; Jeong, J.H.; Redmond, C.K. Breast cancer adjuvant therapy: Time to consider its time-dependent effects. J. Clin. Oncol. 2011, 29, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.D.; Jiang, Y.Z.; Chen, S.; Cao, Z.G.; Wu, J.; Shen, Z.Z.; Shao, Z.M. Effect of large tumor size on cancer-specific mortality in node-negative breast cancer. Mayo Clin. Proc. 2012, 87, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.G.; Dickler, M.N. Endocrine resistance in hormone-responsive breast cancer: Mechanisms and therapeutic strategies. Endocr. Relat. Cancer 2016, 23, R337–R352. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Nardone, A.; De Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The changing role of ER in endocrine resistance. Breast 2015, 24 (Suppl. S2), S60–S66. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef] [Green Version]
- Massarweh, S.; Schiff, R. Resistance to endocrine therapy in breast cancer: Exploiting estrogen receptor/growth factor signaling crosstalk. Endocr. Relat. Cancer 2006, 13 (Suppl. S1), S15–S24. [Google Scholar] [CrossRef]
- Giuliano, M.; Trivedi, M.V.; Schiff, R. Bidirectional Crosstalk between the Estrogen Receptor and Human Epidermal Growth Factor Receptor 2 Signaling Pathways in Breast Cancer: Molecular Basis and Clinical Implications. Breast Care 2013, 8, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef] [Green Version]
- Gates, L.A.; Gu, G.; Chen, Y.; Rohira, A.D.; Lei, J.T.; Hamilton, R.A.; Yu, Y.; Lonard, D.M.; Wang, J.; Wang, S.P.; et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene 2018, 37, 4581–4598. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Hutchinson, K.E.; Stricker, T.P.; Formisano, L.; Young, C.D.; Estrada, M.V.; Nixon, M.J.; Du, L.; Sanchez, V.; Ericsson, P.G.; et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med. 2017, 9, eaai7993. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.J.; Sanchez, B.C.; Forshed, J.; Stal, O.; Fohlin, H.; Lewensohn, R.; Hall, P.; Bergh, J.; Lehtio, J.; Linderholm, B.K. Proteomics profiling identify CAPS as a potential predictive marker of tamoxifen resistance in estrogen receptor positive breast cancer. Clin. Proteom. 2015, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Muller, B.; Brockmoller, S.; Seppanen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Zhou, B.; Su, M.; Baxter, S.; Zheng, X.; Zhao, X.; Yen, Y.; Jia, W. Mass spectrometry-based quantitative metabolomics revealed a distinct lipid profile in breast cancer patients. Int. J. Mol. Sci. 2013, 14, 8047–8061. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Min, H.K.; Kong, G.; Moon, M.H. Quantitative analysis of phosphatidylcholines and phosphatidylethanolamines in urine of patients with breast cancer by nanoflow liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 393, 1649–1656. [Google Scholar] [CrossRef]
- Iwano, T.; Yoshimura, K.; Inoue, S.; Odate, T.; Ogata, K.; Funatsu, S.; Tanihata, H.; Kondo, T.; Ichikawa, D.; Takeda, S. Breast cancer diagnosis based on lipid profiling by probe electrospray ionization mass spectrometry. Br. J. Surg. 2020, 107, 632–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laisupasin, P.; Thompat, W.; Sukarayodhin, S.; Sornprom, A.; Sudjaroen, Y. Comparison of Serum Lipid Profiles between Normal Controls and Breast Cancer Patients. J. Lab. Physicians 2013, 5, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, L.; Liu, N.; He, C.; Li, Z. Decreased serum levels of free fatty acids are associated with breast cancer. Clin. Chim. Acta 2014, 437, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, Y.; Masaki, N.; Takei, S.; Horikawa, M.; Matsushita, S.; Sugiyama, E.; Ogura, H.; Shiiya, N.; Setou, M. Recurrent triple-negative breast cancer (TNBC) tissues contain a higher amount of phosphatidylcholine (32:1) than non-recurrent TNBC tissues. PLoS ONE 2017, 12, e0183724. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A. The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 1994, 269, 3125–3128. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Kronke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef]
- Okazaki, T.; Kondo, T.; Kitano, T.; Tashima, M. Diversity and complexity of ceramide signalling in apoptosis. Cell. Signal. 1998, 10, 685–692. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Xia, Q.S.; Lu, F.E.; Wu, F.; Huang, Z.Y.; Dong, H.; Xu, L.J.; Gong, J. New role for ceramide in hypoxia and insulin resistance. World J. Gastroenterol. 2020, 26, 2177–2186. [Google Scholar] [CrossRef]
- Olivera, A.; Rivera, J. Sphingolipids and the balancing of immune cell function: Lessons from the mast cell. J. Immunol. 2005, 174, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.L.; Fu, P.; Suryadevara, V.; Zhao, Y.; Natarajan, V. Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: Role of S1P lyase. Adv. Biol. Regul. 2017, 63, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnick, R.; Hannun, Y.A. Ceramide and apoptosis. Trends Biochem. Sci. 1999, 24, 224–225; author reply 227. [Google Scholar] [CrossRef]
- Hofmann, K.; Dixit, V.M. Ceramide in apoptosis--does it really matter? Trends Biochem. Sci. 1998, 23, 374–377. [Google Scholar] [CrossRef]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strelow, A.; Bernardo, K.; Adam-Klages, S.; Linke, T.; Sandhoff, K.; Kronke, M.; Adam, D. Overexpression of acid ceramidase protects from tumor necrosis factor-induced cell death. J. Exp. Med. 2000, 192, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Seelan, R.S.; Qian, C.; Yokomizo, A.; Bostwick, D.G.; Smith, D.I.; Liu, W. Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes Cancer 2000, 29, 137–146. [Google Scholar] [CrossRef]
- Bourteele, S.; Hausser, A.; Doppler, H.; Horn-Muller, J.; Ropke, C.; Schwarzmann, G.; Pfizenmaier, K.; Muller, G. Tumor necrosis factor induces ceramide oscillations and negatively controls sphingolipid synthases by caspases in apoptotic Kym-1 cells. J. Biol. Chem. 1998, 273, 31245–31251. [Google Scholar] [CrossRef] [Green Version]
- Maczis, M.A.; Maceyka, M.; Waters, M.R.; Newton, J.; Singh, M.; Rigsby, M.F.; Turner, T.H.; Alzubi, M.A.; Harrell, J.C.; Milstien, S.; et al. Sphingosine kinase 1 activation by estrogen receptor alpha36 contributes to tamoxifen resistance in breast cancer. J. Lipid Res. 2018, 59, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [Green Version]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Gutgesell, L.M.; Xiong, R.; Zhao, J.; Li, Y.; Rosales, C.I.; Hollas, M.; Shen, Z.; Gordon-Blake, J.; Dye, K.; et al. Design and Synthesis of Basic Selective Estrogen Receptor Degraders for Endocrine Therapy Resistant Breast Cancer. J. Med. Chem. 2019, 62, 11301–11323. [Google Scholar] [CrossRef] [PubMed]
- Semina, S.E.; Pal, P.; Kansara, N.S.; Huggins, R.J.; Alarid, E.T.; Greene, G.L.; Frasor, J. Selective pressure of endocrine therapy activates the integrated stress response through NFkappaB signaling in a subpopulation of ER positive breast cancer cells. Breast Cancer Res. 2022, 24, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Fong, Y.; Tsai, E.M.; Chang, Y.G.; Chou, H.L.; Wu, C.Y.; Teng, Y.N.; Liu, T.C.; Yuan, S.S.; Chiu, C.C. Exogenous C(8)-Ceramide Induces Apoptosis by Overproduction of ROS and the Switch of Superoxide Dismutases SOD1 to SOD2 in Human Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesinghe, D.S.; Subramanian, P.; Lamour, N.F.; Gentile, L.B.; Granado, M.H.; Bielawska, A.; Szulc, Z.; Gomez-Munoz, A.; Chalfant, C.E. Chain length specificity for activation of cPLA2alpha by C1P: Use of the dodecane delivery system to determine lipid-specific effects. J. Lipid Res. 2009, 50, 1986–1995. [Google Scholar] [CrossRef] [Green Version]
- Millner, A.; Lizardo, D.Y.; Atilla-Gokcumen, G.E. Untargeted Lipidomics Highlight the Depletion of Deoxyceramides during Therapy-Induced Senescence. Proteomics 2020, 20, e2000013. [Google Scholar] [CrossRef]
- Millner, A.; Running, L.; Colon-Rosa, N.; Aga, D.S.; Frasor, J.; Atilla-Gokcumen, G.E. Ceramide-1-Phosphate Is Involved in Therapy-Induced Senescence. ACS Chem. Biol. 2022, 17, 822–828. [Google Scholar] [CrossRef]
- Smart, E.; Alejo, L.H.; Frasor, J. Cytoplasmic ER alpha and NF kappa B Promote Cell Survival in Mouse Mammary Cancer Cell Lines. Horm. Cancer 2020, 11, 76–86. [Google Scholar] [CrossRef]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Gyorffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Graf, C.; Klumpp, M.; Habig, M.; Rovina, P.; Billich, A.; Baumruker, T.; Oberhauser, B.; Bornancin, F. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol. Pharmacol. 2008, 74, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.-H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gomez-Munoz, A. Ceramide 1-phosphate inhibits serine palmitoyltransferase and blocks apoptosis in alveolar macrophages. Biochim. Biophys. Acta 2009, 1791, 263–272. [Google Scholar] [CrossRef]
- Perry, D.K.; Carton, J.; Shah, A.K.; Meredith, F.; Uhlinger, D.J.; Hannun, Y.A. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J. Biol. Chem. 2000, 275, 9078–9084. [Google Scholar] [CrossRef] [Green Version]
- Hakomori, S.I. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth, adhesion, and motility. FEBS Lett. 2010, 584, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Liu, Y.Y. Sphingolipids and expression regulation of genes in cancer. Prog. Lipid Res. 2011, 50, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.; Bao, J.; Mehendale, H.; Cabot, M.C.; Li, Y.T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol. Cancer 2010, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.P.; Maurer, B.J.; Kolesnick, R.N. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett. 2004, 206, 169–180. [Google Scholar] [CrossRef]
- Siskind, L.J.; Colombini, M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 2000, 275, 38640–38644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Muller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Muriel, M.P.; Lambeng, N.; Darios, F.; Michel, P.P.; Hirsch, E.C.; Agid, Y.; Ruberg, M. Mitochondrial free calcium levels (Rhod-2 fluorescence) and ultrastructural alterations in neuronally differentiated PC12 cells during ceramide-dependent cell death. J. Comp. Neurol. 2000, 426, 297–315. [Google Scholar] [CrossRef]
- Arora, A.S.; Jones, B.J.; Patel, T.C.; Bronk, S.F.; Gores, G.J. Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology 1997, 25, 958–963. [Google Scholar] [CrossRef]
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef]
- Morad, S.A.F.; MacDougall, M.R.; Abdelmageed, N.; Kao, L.P.; Feith, D.J.; Tan, S.F.; Kester, M.; Loughran, T.P., Jr.; Wang, H.G.; Cabot, M.C. Pivotal role of mitophagy in response of acute myelogenous leukemia to a ceramide-tamoxifen-containing drug regimen. Exp. Cell Res. 2019, 381, 256–264. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Klein, S.D.; Schucht, O.; Schenk, U.; Pruschy, M.; Rocha, S.; Richter, C. Ceramide induces cytochrome c release from isolated mitochondria. Importance of mitochondrial redox state. J. Biol. Chem. 1999, 274, 6080–6084. [Google Scholar] [CrossRef] [Green Version]
- Geley, S.; Hartmann, B.L.; Kofler, R. Ceramides induce a form of apoptosis in human acute lymphoblastic leukemia cells that is inhibited by Bcl-2, but not by CrmA. FEBS Lett. 1997, 400, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, D.A.; Kilkus, J.P.; Gottschalk, A.R.; Quintans, J.; Dawson, G. Anti-immunoglobulin-induced apoptosis in WEHI 231 cells involves the slow formation of ceramide from sphingomyelin and is blocked by bcl-XL. J. Biol. Chem. 1997, 272, 9868–9876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [Green Version]
- Hasson, S.P.; Rubinek, T.; Ryvo, L.; Wolf, I. Endocrine resistance in breast cancer: Focus on the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin signaling pathway. Breast Care 2013, 8, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, W.; Fan, P.; Wang, J.; Li, Y.; Santen, R.J. Mechanisms of acquired resistance to endocrine therapy in hormone-dependent breast cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 106, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; You, L.; Lu, Y.; Han, S.; Wang, J.; Vicas, N.; Chen, C.; Ye, J. Identification of TRAMs as sphingolipid-binding proteins using a photoactivatable and clickable short-chain ceramide analog. J. Biol. Chem. 2021, 297, 101415. [Google Scholar] [CrossRef]
- Pulkoski-Gross, M.J.; Donaldson, J.C.; Obeid, L.M. Sphingosine-1-phosphate metabolism: A structural perspective. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Ruckhaberle, E.; Karn, T.; Rody, A.; Hanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1005–1013. [Google Scholar] [CrossRef]
- Payne, A.W.; Pant, D.K.; Pan, T.C.; Chodosh, L.A. Ceramide kinase promotes tumor cell survival and mammary tumor recurrence. Cancer Res. 2014, 74, 6352–6363. [Google Scholar] [CrossRef] [Green Version]
- Pettus, B.J.; Bielawska, A.; Subramanian, P.; Wijesinghe, D.S.; Maceyka, M.; Leslie, C.C.; Evans, J.H.; Freiberg, J.; Roddy, P.; Hannun, Y.A.; et al. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004, 279, 11320–11326. [Google Scholar] [CrossRef] [Green Version]
- Gangoiti, P.; Arana, L.; Ouro, A.; Granado, M.H.; Trueba, M.; Gomez-Munoz, A. Activation of mTOR and RhoA is a major mechanism by which Ceramide 1-phosphate stimulates macrophage proliferation. Cell. Signal. 2011, 23, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gomez-Munoz, A. Activation of protein kinase C-alpha is essential for stimulation of cell proliferation by ceramide 1-phosphate. FEBS Lett. 2010, 584, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangoiti, P.; Granado, M.H.; Wang, S.W.; Kong, J.Y.; Steinbrecher, U.P.; Gomez-Munoz, A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3-kinase/PKB, JNK and ERK1/2 pathways. Cell. Signal. 2008, 20, 726–736. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, P.; Millner, A.; Semina, S.E.; Huggins, R.J.; Running, L.; Aga, D.S.; Tonetti, D.A.; Schiff, R.; Greene, G.L.; Atilla-Gokcumen, G.E.; et al. Endocrine Therapy-Resistant Breast Cancer Cells Are More Sensitive to Ceramide Kinase Inhibition and Elevated Ceramide Levels Than Therapy-Sensitive Breast Cancer Cells. Cancers 2022, 14, 2380. https://doi.org/10.3390/cancers14102380
Pal P, Millner A, Semina SE, Huggins RJ, Running L, Aga DS, Tonetti DA, Schiff R, Greene GL, Atilla-Gokcumen GE, et al. Endocrine Therapy-Resistant Breast Cancer Cells Are More Sensitive to Ceramide Kinase Inhibition and Elevated Ceramide Levels Than Therapy-Sensitive Breast Cancer Cells. Cancers. 2022; 14(10):2380. https://doi.org/10.3390/cancers14102380
Chicago/Turabian StylePal, Purab, Alec Millner, Svetlana E. Semina, Rosemary J. Huggins, Logan Running, Diana S. Aga, Debra A. Tonetti, Rachel Schiff, Geoffrey L. Greene, G. Ekin Atilla-Gokcumen, and et al. 2022. "Endocrine Therapy-Resistant Breast Cancer Cells Are More Sensitive to Ceramide Kinase Inhibition and Elevated Ceramide Levels Than Therapy-Sensitive Breast Cancer Cells" Cancers 14, no. 10: 2380. https://doi.org/10.3390/cancers14102380