
Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers

Abstract
:Simple Summary
Abstract
1. Introduction
2. Analytical Techniques in Post-Translational Modification Analysis
2.1. Phosphorylation
2.2. Ubiquitination
2.3. Glycosylation
2.4. Sumoylation
2.5. Acetylation and Methylation
3. PTM Crosstalk
4. Application of PTM-Focused Techniques in Blood Cancer Research
5. Multiple Myeloma
6. Acute Myeloid Leukemia
7. Myeloproliferative Neoplasms
8. Lymphomas
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakubu, R.R.; Nieves, E.; Weiss, L.M. The Methods Employed in Mass Spectrometric Analysis of Posttranslational Modifications (PTMs) and Protein–Protein Interactions (PPIs). Adv. Exp. Med. Biol. 2019, 1140, 169–198. [Google Scholar] [CrossRef]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Mansour, M.R. The role of noncoding mutations in blood cancers. Dis. Model. Mech. 2019, 12, dmm041988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulte, D.; Jansen, L.; Brenner, H. Changes in long term survival after diagnosis with common hematologic malignancies in the early 21st century. Blood Cancer J. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Gnad, F.; Mann, M. The Case for Proteomics and Phospho-Proteomics in Personalized Cancer Medicine. Proteom. Clin. Appl. 2019, 13, e1800113. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R. Filter Aided Sample Preparation—A tutorial. Anal. Chim. Acta 2019, 1090, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Mann, M. A beginner’s guide to mass spectrometry–based proteomics. Biochemist 2020, 42, 64–69. [Google Scholar] [CrossRef]
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. BioTechniques 2006, 40, 790–798. [Google Scholar] [CrossRef]
- Siuzdak, G. An introduction to mass spectrometry ionization: An excerpt from The Expanding Role of Mass Spectrometry in Biotechnology, 2nd ed.; MCC Press: San Diego, 2005. J. Lab. Autom. 2004, 9, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Fort, K.L.; Cramer, C.N.; Voinov, V.G.; Vasil’Ev, Y.V.; Lopez, N.I.; Beckman, J.S.; Heck, A.J.R. Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer. J. Proteome Res. 2017, 17, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Penkert, M.; Hauser, A.; Harmel, R.; Fiedler, D.; Hackenberger, C.P.R.; Krause, E. Electron Transfer/Higher Energy Collisional Dissociation of Doubly Charged Peptide Ions: Identification of Labile Protein Phosphorylations. J. Am. Soc. Mass Spectrom. 2019, 30, 1578–1585. [Google Scholar] [CrossRef]
- Frese, C.K.; Zhou, H.; Taus, T.; Altelaar, A.F.M.; Mechtler, K.; Heck, A.J.R.; Mohammed, S. Unambiguous Phosphosite Localization using Electron-Transfer/Higher-Energy Collision Dissociation (EThcD). J. Proteome Res. 2013, 12, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Hebert, A.S.; Dürnberger, G.; Stanek, F.; Mechtler, K.; Westphall, M.S.; Coon, J.J. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal. Chem. 2017, 89, 6367–6376. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Wang, B.; Chen, Z.; Urabe, G.; Glover, M.S.; Shi, X.; Guo, L.-W.; Kent, K.C.; Li, L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Chalkley, R.J.; Clauser, K.R. Modification Site Localization Scoring: Strategies and Performance. Mol. Cell. Proteom. 2012, 11, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megger, D.A.; Pott, L.L.; Ahrens, M.; Padden, J.; Bracht, T.; Kuhlmann, K.; Eisenacher, M.; Meyer, H.E.; Sitek, B. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta (BBA)Proteins Proteom. 2014, 1844, 967–976. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S. Label-Based and Label-Free Strategies for Protein Quantitation. Adv. Struct. Saf. Stud. 2016, 1549, 31–43. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Nemutlu, E.; Zhang, S.; Gupta, A.; Juranic, N.O.; Macura, S.I.; Terzic, A.; Jahangir, A.; Dzeja, P.P. Dynamic phosphometabolomic profiling of human tissues and transgenic models by18O-assisted31P NMR and mass spectrometry. Physiol. Genom. 2012, 44, 386–402. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Zang, Q.; Lv, Q.; Gao, Y.; Zhao, Z.; He, J.; Zhang, R.; Song, Y.; Chen, Y.; Abliz, Z. Development of methionine methylation profiling and relative quantification in human breast cancer cells based on metabolic stable isotope labeling. Analyst 2019, 144, 3988–3998. [Google Scholar] [CrossRef]
- Lund, P.J.; Kori, Y.; Zhao, X.; Sidoli, S.; Yuan, Z.-F.; Garcia, B.A. Isotopic Labeling and Quantitative Proteomics of Acetylation on Histones and Beyond. Breast Cancer 2019, 1977, 43–70. [Google Scholar] [CrossRef]
- Filiou, M.D.; Martins-De-Souza, D.; Guest, P.C.; Bahn, S.; Turck, C.W. To label or not to label: Applications of quantitative proteomics in neuroscience research. Proteomics 2012, 12, 736–747. [Google Scholar] [CrossRef]
- Refsgaard, J.C.; Munk, S.; Jensen, L.J. Search Databases and Statistics: Pitfalls and Best Practices in Phosphoproteomics. Adv. Struct. Safety Stud. 2016, 1355, 323–339. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.-Y.; Jiang, Y.; Li, C.-Q.; Zhang, Y.; Dakle, P.; Kaur, H.; Deng, J.-W.; Lin, R.Y.-T.; Han, L.; Xie, J.-J.; et al. TP63, SOX2, and KLF5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology 2020, 159, 1311–1327.e19. [Google Scholar] [CrossRef]
- Kim, S.W.; Hasanuzzaman, M.; Cho, M.; Heo, Y.R.; Ryu, M.-J.; Ha, N.-Y.; Park, H.J.; Park, H.-Y.; Shin, J.-G. Casein Kinase 2 (CK2)-mediated Phosphorylation of Hsp90β as a Novel Mechanism of Rifampin-induced MDR1 Expression. J. Biol. Chem. 2015, 290, 17029–17040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kruijsbergen, I.; Mulder, M.P.C.; Uckelmann, M.; Van Welsem, T.; De Widt, J.; Spanjaard, A.; Jacobs, H.; El Oualid, F.; Ovaa, H.; Van Leeuwen, F. Strategy for Development of Site-Specific Ubiquitin Antibodies. Front. Chem. 2020, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeney, M.; Hedley, B.D.; Chin-Yee, I.H. Flow cytometry-Recognizing unusual populations in leukemia and lymphoma diagnosis. Int. J. Lab. Hematol. 2017, 39, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.-K.; Noh, E.-K.; Ju, L.J.; Sung, J.Y.; Jeong, Y.K.; Cheon, J.; Koh, S.J.; Min, Y.J.; Choi, Y.; Jo, J.-C. CD45dimCD34+CD38−CD133+ cells have the potential as leukemic stem cells in acute myeloid leukemia. BMC Cancer 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Ngai, L.L.; Kelder, A.; Janssen, J.J.W.M.; Ossenkoppele, G.J.; Cloos, J. MRD Tailored Therapy in AML: What We Have Learned So Far. Front. Oncol. 2021, 10, 603636. [Google Scholar] [CrossRef]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, J.; Liu, J.; Xie, Y.; Li, X.; Jiang, H.; Wang, J.; Peng, Z.; Wang, J.; Wang, S.; et al. Hypoxia-induced CREB cooperates MMSET to modify chromatin and promote DKK1 expression in multiple myeloma. Oncogene 2021, 40, 1231–1241. [Google Scholar] [CrossRef]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A.; et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2010, 18, 91–93. [Google Scholar] [CrossRef] [Green Version]
- Byrum, S.D.; Taverna, S.D.; Tackett, A.J. Purification of specific chromatin loci for proteomic analysis. Breast Cancer 2014, 1228, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Sidoli, S.; Bhanu, N.V.; Karch, K.R.; Wang, X.; Garcia, B.A. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J. Vis. Exp. 2016, e54112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, L.D.; Steger, M.G.; Polyansky, A.A.; Schubert, T.; Zagrovic, B.; Zheng, N.; Clausen, T.; Herzog, F.; Köhler, A. Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6–Bre1. Proc. Natl. Acad. Sci. USA 2016, 113, 10553–10558. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Riedl, T.; Egly, J.-M. Phosphorylation in Transcription: The CTD and More. Gene Expr. 2001, 9, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and Downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The Oxidative Phosphorylation System in Mammalian Mitochondria. Adv. Exp. Med. Biol. 2011, 942, 3–37. [Google Scholar] [CrossRef]
- VerPlank, J.J.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef] [PubMed]
- Natoli, C.; Perrucci, B.; Perrotti, F.; Falchi, L.; Iacobelli, S. Tyrosine Kinase Inhibitors. Curr. Cancer Drug Targets 2010, 10, 462–483. [Google Scholar] [CrossRef] [PubMed]
- Bodenmiller, B.; Mueller, L.N.; Mueller, M.; Domon, B.; Aebersold, R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat. Chem. Biol. 2007, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E.; Rogowska-Wrzesinska, A. The challenge of detecting modifications on proteins. Essays Biochem. 2020, 64, 135–153. [Google Scholar] [CrossRef]
- Ke, M.; Shen, H.; Wang, L.; Luo, S.; Lin, L.; Yang, J.; Tian, R. Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics. Adv. Exp. Med. Biol. 2016, 919, 345–382. [Google Scholar] [CrossRef]
- Raggiaschi, R.; Gotta, S.; Terstappen, G.C. Phosphoproteome Analysis. Biosci. Rep. 2005, 25, 33–44. [Google Scholar] [CrossRef]
- Zavialova, M.; Zgoda, V.; Nikolaev, E. Analysis of contribution of protein phosphorylation in the development of the diseases. Biomeditsinskaya Khimiya 2017, 63, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Sawasdikosol, S. Detecting Tyrosine-Phosphorylated Proteins by Western Blot Analysis. Curr. Protoc. Immunol. 2010, 89, 11.3.1–11.3.11. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, X.; Pelech, S. Monitoring Protein Kinase Expression and Phosphorylation in Cell Lysates with Antibody Microarrays. Adv. Struct. Saf. Stud. 2016, 1360, 107–122. [Google Scholar] [CrossRef]
- Pierobon, M.; Wulfkuhle, J.; Liotta, L.A.; Iii, E.F.P. Utilization of Proteomic Technologies for Precision Oncology Applications. Cancer Treat. Res. 2019, 178, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pelech, S. Using protein microarrays to study phosphorylation-mediated signal transduction. Semin. Cell Dev. Biol. 2012, 23, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Kyr, M.; Polaskova, K.; Kren, L.; Macigova, P.; Skoda, J.; Sterba, J.; Veselska, R. Phospho-Protein Arrays as Effec-tive Tools for Screening Possible Targets for Kinase Inhibitors and Their Use in Precision Pediatric Oncology. Front. Cology 2019, 9, 930. [Google Scholar]
- Sukswai, N.; Khoury, J.D. Immunohistochemistry Innovations for Diagnosis and Tissue-Based Biomarker Detection. Curr. Hematol. Malign Rep. 2019, 14, 368–375. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Trejo, A.; Schulz, K.R.; Nolan, G.P. Phospho Flow Cytometry Methods for the Analysis of Kinase Signaling in Cell Lines and Primary Human Blood Samples. Methods Mol. Biol. 2010, 699, 179–202. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villén, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [Green Version]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, B.; Koch, H.; Medard, G.; Mundt, M.; Kuster, B.; Lemeer, S. Comprehensive and Reproducible Phosphopeptide Enrichment Using Iron Immobilized Metal Ion Affinity Chromatography (Fe-IMAC) Columns. Mol. Cell. Proteom. 2015, 14, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantin, G.T.; Shock, T.R.; Park, S.K.; Madhani, H.D.; Yates, J.R. Optimizing TiO2-Based Phosphopeptide Enrichment for Automated Multidimensional Liquid Chromatography Coupled to Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 4666–4673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, S.; León, I.R.; Jensen, O.N.; Højlund, K. Tissue Specific Phosphorylation of Mitochondrial Proteins Isolated from Rat Liver, Heart Muscle, and Skeletal Muscle. J. Proteome Res. 2013, 12, 4327–4339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Watts, J.D.; Aebersold, R. A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 2001, 19, 375–378. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y. β-Elimination Coupled with Tandem Mass Spectrometry for the Identification of in Vivo and in Vitro Phosphorylation Sites in Maize Dehydrin DHN1 Protein. Biochemistry 2004, 43, 15567–15576. [Google Scholar] [CrossRef]
- Edelmann, M.J. Strong Cation Exchange Chromatography in Analysis of Posttranslational Modifications: Innovations and Perspectives. J. Biomed. Biotechnol. 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Mohammed, S.; Heck, A.J. Strong cation exchange (SCX) based analytical methods for the targeted analysis of protein post-translational modifications. Curr. Opin. Biotechnol. 2011, 22, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Boersema, P.J.; Mohammed, S.; Heck, A.J.R. Hydrophilic interaction liquid chromatography (HILIC) in proteomics. Anal. Bioanal. Chem. 2008, 391, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpert, A.J. Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Bodenmiller, B.; Aebersold, R. Quantitative Analysis of Protein Phosphorylation on a System-Wide Scale by Mass Spectrometry-Based Proteomics. Methods Enzymol. 2010, 470, 317–334. [Google Scholar] [CrossRef]
- Leitner, A. Enrichment Strategies in Phosphoproteomics. Methods Mol. Biol. 2016, 1355, 105–121. [Google Scholar] [CrossRef]
- Zhou, H.; Ye, M.; Dong, J.; Han, G.; Jiang, X.; Wu, R.; Zou, H. Specific Phosphopeptide Enrichment with Immobilized Titanium Ion Affinity Chromatography Adsorbent for Phosphoproteome Analysis. J. Proteome Res. 2008, 7, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Iliuk, A.B.; Martin, V.A.; Alicie, B.M.; Geahlen, R.L.; Tao, W.A. In-depth Analyses of Kinase-dependent Tyrosine Phosphoproteomes Based on Metal Ion-functionalized Soluble Nanopolymers. Mol. Cell. Proteom. 2010, 9, 2162–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasundera, K.B.; Iliuk, A.B.; Nguyen, A.; Higgins, R.; Geahlen, R.L.; Tao, W.A. Global Phosphoproteomics of Activated B Cells Using Complementary Metal Ion Functionalized Soluble Nanopolymers. Anal. Chem. 2014, 86, 6363–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Jie, J.; Yang, B. Single-Step Enrichment of N-Glycopeptides and Phosphopeptides with Novel Multifunctional Ti4+-Immobilized Dendritic Polyglycerol Coated Chitosan Nanomaterials. Anal. Chem. 2017, 89, 7520–7526. [Google Scholar] [CrossRef] [PubMed]
- Díez, I.A.; Govender, I.; Naicker, P.; Stoychev, S.; Jordaan, J.; Jensen, O.N. Zirconium(IV)-IMAC Revisited: Improved Performance and Phosphoproteome Coverage by Magnetic Microparticles for Phosphopeptide Affinity Enrichment. J. Proteome Res. 2021, 20, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.D.; Reid, G.E.; Bruening, M.L. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom. Rev. 2009, 29, 29–54. [Google Scholar] [CrossRef]
- Engholm-Keller, K.; Larsen, M.R. Titanium dioxide as chemo-affinity chromatographic sorbent of biomolecular compounds—Applications in acidic modification-specific proteomics. J. Proteom. 2011, 75, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Carrera, M.; Cañas, B.; Lopez-Ferrer, D. Fast Global Phosphoproteome Profiling of Jurkat T Cells by HIFU-TiO2-SCX-LC-MS/MS. Anal. Chem. 2017, 89, 8853–8862. [Google Scholar] [CrossRef]
- Junemann, J.; Just, I.; Gerhard, R.; Pich, A. Quantitative Phosphoproteome Analysis of Clostridioides difficile Toxin B Treated Human Epithelial Cells. Front. Microbiol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Zhan, Q.; Zheng, Y.; Pu, C.; Zhao, H.; Lan, M. Hydrophilic phytic acid-functionalized magnetic dendritic mesoporous silica nanospheres with immobilized Ti4+: A dual-purpose affinity material for highly efficient enrichment of glycopeptides/phosphopeptides. Talanta 2019, 197, 77–85. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jensen, O.N.; Robinson, P.J.; Larsen, M.R. SIMAC (Sequential Elution from IMAC), a Phosphoproteomics Strategy for the Rapid Separation of Monophosphorylated from Multiply Phosphorylated Peptides. Mol. Cell. Proteom. 2008, 7, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engholm-Keller, K.; Larsen, M.R. Improving the Phosphoproteome Coverage for Limited Sample Amounts Using TiO2-SIMAC-HILIC (TiSH) Phosphopeptide Enrichment and Fractionation. Adv. Struct. Saf. Stud. 2016, 1355, 161–177. [Google Scholar] [CrossRef]
- Gropengiesser, J.; Varadarajan, B.T.; Stephanowitz, H.; Krause, E. The relative influence of phosphorylation and methylation on responsiveness of peptides to MALDI and ESI mass spectrometry. J. Mass Spectrom. 2009, 44, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Jebanathirajah, J.A.; Rush, J.; Morrice, N.; Kirschner, M.W. Phosphorylation Analysis by Mass Spectrometry. Mol. Cell. Proteom. 2006, 5, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Tiambeng, T.N.; Cai, W.; Chen, B.; Lin, Z.; Gregorich, Z.R.; Ge, Y. Impact of Phosphorylation on the Mass Spectrometry Quantification of Intact Phosphoproteins. Anal. Chem. 2018, 90, 4935–4939. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.M.; Vitorino, R.; Domingues, M.M.; Spickett, C.M.; Domingues, P. Post-translational Modifications and Mass Spectrometry Detection. Free. Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef] [Green Version]
- Solari, F.A.; Dell’Aica, M.; Sickmann, A.; Zahedi, R.P. Why phosphoproteomics is still a challenge. Mol. BioSyst. 2015, 11, 1487–1493. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Ong, S.-E.; Grønborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Tichy, A.; Salovska, B.; Rehulka, P.; Klimentova, J.; Vavrova, J.; Stulik, J.; Hernychova, L. Phosphoproteomics: Searching for a needle in a haystack. J. Proteom. 2011, 74, 2786–2797. [Google Scholar] [CrossRef]
- Rose, A.; Mayor, T. Exploring the Rampant Expansion of Ubiquitin Proteomics. Methods Mol. Biol. 2018, 1844, 345–362. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulzele, A.; Bennett, E.J. Ubiquitin diGLY Proteomics as an Approach to Identify and Quantify the Ubiquitin-Modified Proteome. Adv. Struct. Saf. Stud. 2018, 1844, 363–384. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsen, J.M.R.; Sylvestersen, K.B.; Bekker-Jensen, S.; Szklarczyk, D.; Poulsen, J.W.; Horn, H.; Jensen, L.J.; Mailand, N.; Nielsen, M.L. Mass Spectrometric Analysis of Lysine Ubiquitylation Reveals Promiscuity at Site Level. Mol. Cell. Proteom. 2011, 10, 110–003590. [Google Scholar] [CrossRef] [Green Version]
- Pontrelli, P.; Conserva, F.; Papale, M.; Oranger, A.; Barozzino, M.; Vocino, G.; Rocchetti, M.T.; Gigante, M.; Castellano, G.; Rossini, M.; et al. Lysine 63 ubiquitination is involved in the progression of tubular damage in diabetic nephropathy. FASEB J. 2017, 31, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doellinger, J.; Grossegesse, M.; Nitsche, A.; Lasch, P. DMSO as a mobile phase additive enhances detection of ubiquitination sites by nano-LC-ESI-MS/MS. J. Mass Spectrom. 2018, 53, 183–187. [Google Scholar] [CrossRef]
- Porras-Yakushi, T.R.; Sweredoski, M.J.; Hess, S. ETD Outperforms CID and HCD in the Analysis of the Ubiquitylated Proteome. J. Am. Soc. Mass Spectrom. 2015, 26, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udeshi, N.D.; Mani, D.C.; Satpathy, S.; Fereshetian, S.; Gasser, J.A.; Svinkina, T.; Olive, M.E.; Ebert, B.L.; Mertins, P.; Carr, S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Harris, L.D.; Le Pen, J.; Scholz, N.; Mieszczanek, J.; Vaughan, N.; Davis, S.; Berridge, G.; Kessler, B.M.; Bienz, M.; Licchesi, J.D. The deubiquitinase TRABID stabilizes the K29/K48-specific E3 ubiquitin ligase HECTD1. J. Biol. Chem. 2021, 296, 100246. [Google Scholar] [CrossRef]
- Ordureau, A.; Münch, C.; Harper, J.W. Quantifying Ubiquitin Signaling. Mol. Cell 2015, 58, 660–676. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Liu, S.; Sagum, C.; Chen, J.; Singh, R.; Chaturvedi, A.; Horton, J.R.; Kashyap, T.R.; Fushman, D.; Cheng, X.; et al. Crosstalk between Lys63- and Lys11-polyubiquitin signaling at DNA damage sites is driven by Cezanne. Genes Dev. 2019, 33, 1702–1717. [Google Scholar] [CrossRef] [PubMed]
- Akinjiyan, F.A.; Fazal, A.; Hild, M.; Beckwith, R.E.J.; Ross, N.T.; Paulk, J.; Carbonneau, S. A Novel Luminescence-Based High-Throughput Approach for Cellular Resolution of Protein Ubiquitination Using Tandem Ubiquitin Binding Entities (TUBEs). SLAS Discov. Adv. Life Sci. R D 2020, 25, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.-J.; Rape, M. Enhanced Protein Degradation by Branched Ubiquitin Chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Crowe, S.O.; Rana, A.S.J.B.; Deol, K.K.; Ge, Y.; Strieter, E.R. Ubiquitin Chain Enrichment Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains in Cellulo. Anal. Chem. 2017, 89, 4428–4434. [Google Scholar] [CrossRef] [Green Version]
- Valkevich, E.M.; Sanchez, N.A.; Ge, Y.; Strieter, E.R. Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains. Biochemistry 2014, 53, 4979–4989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-S.; Wu, K.-P.; Jiang, H.-K.; Kurkute, P.; Chen, R.-H. Branched Ubiquitination: Detection Methods, Biological Functions and Chemical Synthesis. Molecules 2020, 25, 5200. [Google Scholar] [CrossRef]
- King, S.L.; Joshi, H.J.; Schjoldager, K.T.; Halim, A.; Madsen, T.D.; Dziegiel, M.H.; Woetmann, A.; Vakhrushev, S.Y.; Wandall, H.H. Characterizing the O-glycosylation landscape of human plasma, platelets, and endothelial cells. Blood Adv. 2017, 1, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Anonsen, J.H.; Vik, Å.; Egge-Jacobsen, W.; Koomey, M. An Extended Spectrum of Target Proteins and Modification Sites in the General O-Linked Protein Glycosylation System in Neisseria gonorrhoeae. J. Proteome Res. 2012, 11, 5781–5793. [Google Scholar] [CrossRef] [PubMed]
- Dan, X.; Liu, W.; Ng, T.B. Development and Applications of Lectins as Biological Tools in Biomedical Research. Med. Res. Rev. 2015, 36, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Durham, M.; Regnier, F.E. Targeted glycoproteomics: Serial lectin affinity chromatography in the selection of O-glycosylation sites on proteins from the human blood proteome. J. Chromatogr. A 2006, 1132, 165–173. [Google Scholar] [CrossRef]
- Totten, S.M.; Adusumilli, R.; Kullolli, M.; Tanimoto, C.; Brooks, J.D.; Mallick, P.; Pitteri, S.J. Multi-lectin Affinity Chromatography and Quantitative Proteomic Analysis Reveal Differential Glycoform Levels between Prostate Cancer and Benign Prostatic Hyperplasia Sera. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totten, S.M.; Kullolli, M.; Pitteri, S.J. Multi-Lectin Affinity Chromatography for Separation, Identification, and Quantitation of Intact Protein Glycoforms in Complex Biological Mixtures. Methods Mol. Biol. 2017, 1550, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Sun, F.; Suttapitugsakul, S.; Wu, R. Global and site-specific analysis of protein glycosylation in complex biological systems with Mass Spectrometry. Mass Spectrom. Rev. 2019, 38, 356–379. [Google Scholar] [CrossRef]
- Bai, H.; Pan, Y.; Qi, L.; Liu, L.; Zhao, X.; Dong, H.; Cheng, X.; Qin, W.; Wang, X. Development a hydrazide-functionalized thermosensitive polymer based homogeneous system for highly efficient N-glycoprotein/glycopeptide enrichment from human plasma exosome. Talanta 2018, 186, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Lipinszki, Z.; Kupihár, Z.; Udvardy, A.; Medzihradszky, K.F. Enrichment of O-GlcNAc Modified Proteins by the Periodate Oxidation−Hydrazide Resin Capture Approach. J. Proteome Res. 2010, 9, 2200–2206. [Google Scholar] [CrossRef] [Green Version]
- Calvano, C.D.; Zambonin, C.G.; Jensen, O.N. Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J. Proteom. 2008, 71, 304–317. [Google Scholar] [CrossRef]
- Woo, C.M.; Iavarone, A.T.; Spiciarich, D.R.; Palaniappan, K.K.; Bertozzi, C.R. Isotope-targeted glycoproteomics (IsoTaG): A mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat. Methods 2015, 12, 561–567. [Google Scholar] [CrossRef]
- Sun, S.; Shah, P.; Eshghi, S.T.; Yang, W.; Trikannad, N.; Yang, S.; Chen, L.; Aiyetan, P.; Höti, N.; Zhang, Z.; et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol. 2016, 34, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Triguero, A.; Cabrera, G.; Royle, L.; Harvey, D.J.; Rudd, P.M.; Dwek, R.A.; Bardor, M.; Lerouge, P.; Cremata, J.A. Chemical and enzymatic N-glycan release comparison for N-glycan profiling of monoclonal antibodies expressed in plants. Anal. Biochem. 2010, 400, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Stavenhagen, K.; Hinneburg, H.; Thaysen-Andersen, M.; Hartmann, L.; Silva, D.V.; Fuchser, J.; Kaspar, S.; Rapp, E.; Seeberger, P.H.; Kolarich, D. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: An evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J. Mass Spectrom. 2013, 48, 627–639. [Google Scholar] [CrossRef]
- Wells, L.; Vosseller, K.; Cole, R.N.; Cronshaw, J.M.; Matunis, M.J.; Hart, G.W. Mapping Sites of O-GlcNAc Modification Using Affinity Tags for Serine and Threonine Post-translational Modifications. Mol. Cell. Proteom. 2002, 1, 791–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steentoft, C.; Vakhrushev, S.Y.; Vester-Christensen, M.B.; Schjoldager, K.T.-B.G.; Kong, Y.; Bennett, E.P.; Mandel, U.; Wandall, H.H.; Levery, S.B.; Clausen, H. Mining the O-glycoproteome using zinc-finger nuclease–glycoengineered SimpleCell lines. Nat. Methods 2011, 8, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Zampronio, C.G.; Creese, A.J.; Cooper, H.J. Higher Energy Collision Dissociation (HCD) Product Ion-Triggered Electron Transfer Dissociation (ETD) Mass Spectrometry for the Analysis of N-Linked Glycoproteins. J. Proteome Res. 2012, 11, 4517–4525. [Google Scholar] [CrossRef]
- Eifler, K.; Vertegaal, A.C. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef] [Green Version]
- Park, I.S.; Han, Y.G.; Chung, H.J.; Jung, Y.W.; Kim, Y.; Kim, H. SUMOylation regulates nuclear localization and stability of TRAIP/RNF206. Biochem. Biophys. Res. Commun. 2016, 470, 881–887. [Google Scholar] [CrossRef]
- Rosonina, E.; Akhter, A.; Dou, Y.; Babu, J.; Theivakadadcham, V.S.S. Regulation of transcription factors by sumoylation. Transcription 2017, 8, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Y.; Tang, B.; Zhang, C.-Y. The strategies for identification and quantification of SUMOylation. Chem. Commun. 2017, 53, 6989–6998. [Google Scholar] [CrossRef]
- Sheng, Z.; Wang, X.; Ma, Y.; Zhang, D.; Yang, Y.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. MS-based strategies for identification of protein SUMOylation modification. Electrophoresis 2019, 40, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Impens, F.; Radoshevich, L.; Cossart, P.; Ribet, D. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, 12432–12437. [Google Scholar] [CrossRef] [Green Version]
- Lamoliatte, F.; Caron, D.; Durette, C.; Mahrouche, L.; Maroui, M.A.; Caron-Lizotte, O.; Bonneil, E.; Chelbi-Alix, M.K.; Thibault, P. Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling. Nat. Commun. 2014, 5, 5409. [Google Scholar] [CrossRef] [Green Version]
- Tatham, M.H.; Rodriguez, M.S.; Xirodimas, D.P.; Hay, R.T. Detection of protein SUMOylation in vivo. Nat. Protoc. 2009, 4, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Galisson, F.; Mahrouche, L.; Courcelles, M.; Bonneil, E.; Meloche, S.; Chelbi-Alix, M.K.; Thibault, P. A Novel Proteomics Approach to Identify SUMOylated Proteins and Their Modification Sites in Human Cells. Mol. Cell. Proteom. 2011, 10, S1–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.-M.; Liu, Z.; Okada, M.; Jang, S.-W.; Liu, X.; Chan, C.-B.; Luo, H.; Ye, K. Ebp1 sumoylation, regulated by TLS/FUS E3 ligase, is required for its anti-proliferative activity. Oncogene 2009, 29, 1017–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubiete-Franco, I.; García-Rodríguez, J.L.; Lopitz-Otsoa, F.; Serrano-Macia, M.; Simon, J.; Fernández-Tussy, P.; Barbier-Torres, L.; Fernández-Ramos, D.; Gutiérrez-De-Juan, V.; De Davalillo, S.L.; et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine 2019, 40, 406–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.; Barysch, S.V.; Karaca, S.; Dittner, C.; Hsiao, H.-H.; Diaz, M.B.; Herzig, S.; Urlaub, H.; Melchior, F. Detecting endogenous SUMO targets in mammalian cells and tissues. Nat. Struct. Mol. Biol. 2013, 20, 525–531. [Google Scholar] [CrossRef]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; D’Souza, R.C.; Chang, J.-G.; Mann, M.; Vertegaal, A.C.O. System-wide identification of wild-type SUMO-2 conjugation sites. Nat. Commun. 2015, 6, 7289. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Tatham, M.H.; Plechanovová, A.; Matic, I.; Garg, A.K.; Hay, R.T. Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 2011, 12, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopitz-Otsoa, F.; Delgado, T.C.; Lachiondo-Ortega, S.; Azkargorta, M.; Elortza, F.; Rodríguez, M.S.; Martínez-Chantar, M.L. SUMO-Binding Entities (SUBEs) as Tools for the Enrichment, Isolation, Identification, and Characterization of the SUMO Proteome in Liver Cancer. J. Vis. Exp. 2019, e60098. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’Souza, R.C.J.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handu, M.; Kaduskar, B.; Ravindranathan, R.; Soory, A.; Giri, R.; Elango, V.B.; Gowda, H.; Ratnaparkhi, G.S. SUMO-Enriched Proteome for Drosophila Innate Immune Response. G3: Genes|Genomes|Genetics 2015, 5, 2137–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Pedrioli, P.G.; Raught, B.; Zhang, X.-D.; Rogers, R.; Aitchison, J.; Matunis, M.; Aebersold, R. Automated identification of SUMOylation sites using mass spectrometry and SUMmOn pattern recognition software. Nat. Chem. Biol. 2006, 3, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Chen, Z.; Gong, Y.-A.; Ying, G. SUMOhydro: A Novel Method for the Prediction of Sumoylation Sites Based on Hydrophobic Properties. PLoS ONE 2012, 7, e39195. [Google Scholar] [CrossRef] [Green Version]
- Dehzangi, A.; López, Y.; Taherzadeh, G.; Sharma, A.; Tsunoda, T. SumSec: Accurate Prediction of Sumoylation Sites Using Predicted Secondary Structure. Molecules 2018, 23, 3260. [Google Scholar] [CrossRef] [Green Version]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteom. 2019, 16, 139–159. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Chen, J.; Chen, L.-F. Methods to detect NF-κB acetylation and methylation. Breast Cancer 2015, 1280, 395–409. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, K.J.; Lewis, J.D. Protein Acetylation in Pathogen Virulence and Host Defense: In Vitro Detection of Protein Acetylation by Radiolabeled Acetyl Coenzyme A. Breast Cancer 2019, 1991, 23–32. [Google Scholar] [CrossRef]
- Hatakeyama, D.; Shoji, M.; Yamayoshi, S.; Yoh, R.; Ohmi, N.; Takenaka, S.; Saitoh, A.; Arakaki, Y.; Masuda, A.; Komatsu, T.; et al. Influenza A virus nucleoprotein is acetylated by histone acetyltransferases PCAF and GCN5. J. Biol. Chem. 2018, 293, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronzoni, S.; Faretta, M.; Ballarini, M.; Pelicci, P.; Minucci, S. New method to detect histone acetylation levels by flow cytometry. Cytom. Part A 2005, 66, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Yucel, N.; Wang, Y.X.; Mai, T.; Porpiglia, E.; Lund, P.J.; Markov, G.; Garcia, B.A.; Bendall, S.C.; Angelo, M.; Blau, H.M. Glucose Metabolism Drives Histone Acetylation Landscape Transitions that Dictate Muscle Stem Cell Function. Cell Rep. 2019, 27, 3939–3955.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uitrakul, S.; Hutton, C.; Veal, G.J.; Jamieson, D. A novel imaging flow cytometry method for the detection of histone H4 acetylation in myeloid cells. Eur. J. Clin. Investig. 2019, 49, e13115. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, C.; Han, Y.; Liu, Z.; Wu, T.; Liu, Y.; Tan, Y.; Cai, X.; Cao, Y.; Wang, B.; et al. Identification of Lysine Acetylation in Mycobacterium abscessus Using LC–MS/MS after Immunoprecipitation. J. Proteome Res. 2016, 15, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Meyer, J.G.; Wei, L.; Ott, M.; Verdin, E. High-Resolution Mass Spectrometry to Identify and Quantify Acetylation Protein Targets. Methods Mol. Biol. 2019, 1983, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Fung, S.-Y.; Xie, G.; Wong, L.-Y.R.; Jin, D.-Y.; Cai, Z. Identification of Lysine Acetylation Sites on MERS-CoV Replicase pp1ab. Mol. Cell. Proteom. 2020, 19, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Goethals, M.; Martens, L.; Van Damme, J.; Staes, A.; Thomas, G.R.; Vandekerckhove, J. Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol. 2003, 21, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Kalvik, T.V.; Starheim, K.K.; Jonckheere, V.; Myklebust, L.M.; Menschaert, G.; Varhaug, J.E.; Gevaert, K.; Arnesen, T. A Role for Human N-alpha Acetyltransferase 30 (Naa30) in Maintaining Mitochondrial Integrity. Mol. Cell. Proteom. 2016, 15, 3361–3372. [Google Scholar] [CrossRef] [Green Version]
- Dinh, T.V.; Bienvenut, W.V.; Linster, E.; Feldman-Salit, A.; Jung, V.A.; Meinnel, T.; Hell, R.; Giglione, C.; Wirtz, M. Molecular identification and functional characterization of the first Nα-acetyltransferase in plastids by global acetylome profiling. Proteomics 2015, 15, 2426–2435. [Google Scholar] [CrossRef] [Green Version]
- Bienvenut, W.V.; Giglione, C.; Meinnel, T. SILProNAQ: A Convenient Approach for Proteome-Wide Analysis of Protein N-Termini and N-Terminal Acetylation Quantitation. Breast Cancer 2017, 1574, 17–34. [Google Scholar] [CrossRef]
- Romanick, S.S.; Ulrich, C.; Schlauch, K.; Hostler, A.; Payne, J.; Woolsey, R.; Quilici, D.; Feng, Y.; Ferguson, B.S. Obesity-mediated regulation of cardiac protein acetylation: Parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.-Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Meng, J.; Liu, X.; Peng, X.; Cheng, Z.; Zhang, F.; Zhang, X. ING5 differentially regulates protein lysine acetylation and promotes p300 autoacetylation. Oncotarget 2017, 9, 1617–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrig, P.M.; Hunziker, P.E.; Zahariev, S.; Pongor, S. Fragmentation pathways of NG-methylated and unmodified arginine residues in peptides studied by ESI-MS/MS and MALDI-MS. J. Am. Soc. Mass Spectrom. 2004, 15, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.Y.; Li, Y.; Wang, Y.; Chen, Y.; Zhao, A.Y.; Qin, J. Complications in the Assignment of 14 and 28 Da Mass Shift Detected by Mass Spectrometry as in Vivo Methylation from Endogenous Proteins. Anal. Chem. 2008, 80, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Afjehi-Sadat, L.; Garcia, B.A. Comprehending dynamic protein methylation with mass spectrometry. Curr. Opin. Chem. Biol. 2013, 17, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlmann, T.; Geoghegan, V.L.; Thomas, B.; Ridlova, G.; Trudgian, D.C.; Acuto, O. A Method for Large-scale Identification of Protein Arginine Methylation. Mol. Cell. Proteom. 2012, 11, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, K.; Ye, M. Strategies for large-scale analysis of non-histone protein methylation by LC-MS/MS. Analyst 2017, 142, 3536–3548. [Google Scholar] [CrossRef]
- Hartel, N.G.; Chew, B.; Qin, J.; Xu, J.; Graham, N.A. Deep Protein Methylation Profiling by Combined Chemical and Immunoaffinity Approaches Reveals Novel PRMT1 Targets. Mol. Cell. Proteom. 2019, 18, 2149–2164. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity Enrichment and Mass Spectrometry Analysis of Protein Methylation. Mol. Cell. Proteom. 2014, 13, 372–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, S.M.; E Moore, K.; Green, E.M.; Martín, G.M.; Gozani, O. Proteome-wide enrichment of proteins modified by lysine methylation. Nat. Protoc. 2013, 9, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Wu, H.; Ismail, H.; Du, S.; Cao, J.; Wang, J.; Ward, T.; Yang, F.; Gui, P.; Ali, M.; et al. Methylation of PLK1 by SET7/9 ensures accurate kinetochore–microtubule dynamics. J. Mol. Cell Biol. 2020, 12, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, Z.; Wang, K.; Wang, Y.; Ye, M. A new chromatographic approach to analyze methylproteome with enhanced lysine methylation identification performance. Anal. Chim. Acta 2019, 1068, 111–119. [Google Scholar] [CrossRef]
- Katsanovskaja, K.; Driver, T.; Pipkorn, R.; Edelson-Averbukh, M. Negative Ion Mode Collision-Induced Dissociation for Analysis of Protein Arginine Methylation. J. Am. Soc. Mass Spectrom. 2019, 30, 1229–1241. [Google Scholar] [CrossRef]
- Ong, S.-E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Chem. Biol. 2004, 1, 119–126. [Google Scholar] [CrossRef]
- Geoghegan, V.; Guo, A.; Trudgian, D.; Thomas, B.; Acuto, O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015, 6, 6758. [Google Scholar] [CrossRef]
- Wang, K.; Ye, M. Enrichment of Methylated Peptides Using an Antibody-free Approach for Global Methylproteomics Analysis. Curr. Protoc. Protein Sci. 2018, 91, 14.18.1–14.18.14. [Google Scholar] [CrossRef]
- Zheng, Y.; Fornelli, L.; Compton, P.D.; Sharma, S.; Canterbury, J.; Mullen, C.; Zabrouskov, V.; Fellers, R.T.; Thomas, P.M.; Licht, J.D.; et al. Unabridged Analysis of Human Histone H3 by Differential Top-Down Mass Spectrometry Reveals Hypermethylated Proteoforms from MMSET/NSD2 Overexpression. Mol. Cell. Proteom. 2016, 15, 776–790. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, A.; Marino, F.; Post, H.; Toorn, H.W.P.V.D.; Mohammed, S.; Heck, A.J.R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Huang, R.; Yuan, L. Crosstalk of intracellular post-translational modifications in cancer. Arch. Biochem. Biophys. 2019, 676, 108138. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer1. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.D.; Gevaert, K.; De Smet, I. Protein Language: Post-Translational Modifications Talking to Each Other. Trends Plant Sci. 2018, 23, 1068–1080. [Google Scholar] [CrossRef]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.A. Ubiquitination: Friend and foe in cancer. Int. J. Biochem. Cell Biol. 2018, 101, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. The Multiple Myeloma Drug Pipeline—2018: A Review of Small Molecules and Their Therapeutic Targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salesse, S.; Verfaillie, C.M. BCR/ABL: From molecular mechanisms of leukemia induction to treatment of chronic myelogenous leukemia. Oncogene 2002, 21, 8547–8559. [Google Scholar] [CrossRef] [Green Version]
- Raedler, L.A. Jakafi (Ruxolitinib): First FDA-Approved Medication for the Treatment of Patients with Polycythemia Vera. Am. Health Drug Benefits 2015, 8, 75–79. [Google Scholar]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Waller, C.F. Imatinib Mesylate. Methods Mol. Biol. 2018, 212, 1–27. [Google Scholar]
- Lindauer, M.; Hochhaus, A. Dasatinib. Methods Mol. Biol. 2018, 212, 29–68. [Google Scholar]
- Sacha, T.; Saglio, G. Nilotinib in the treatment of chronic myeloid leukemia. Futur. Oncol. 2019, 15, 953–965. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Massaro, F.; Molica, M.; Breccia, M.; Massaro, M.M.A.M.B.F. Ponatinib: A Review of Efficacy and Safety. Curr. Cancer Drug Targets 2018, 18, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, A.; Schwarzbich, M.-A.; Witzens-Harig, M. Ibrutinib. Methods Mol. Biol. 2018, 212, 133–168. [Google Scholar]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92. [Google Scholar] [CrossRef] [Green Version]
- Groen, K.; Van De Donk, N.W.C.J.; Stege, C.; Zweegman, S.; Nijhof, I. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 2019, ume 11, 2663–2675. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Zweegman, S.; O’Donnell, E.K.; Laubach, J.P.; Raje, N.; Voorhees, P.; Ferrari, R.H.; Skacel, T.; Kumar, S.K.; Lonial, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2018, 19, 1949–1968. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Futur. Oncol. 2017, 13, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat—An Overview. Indian J. Derm. 2015, 60, 419. [Google Scholar]
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Futur. Oncol. 2015, 11, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Smolewski, P.; Robak, T. The discovery and development of romidepsin for the treatment of T-cell lymphoma. Expert Opin. Drug Discov. 2017, 12, 1–15. [Google Scholar] [CrossRef]
- D’Agostino, M.; Bertamini, L.; Oliva, S.; Boccadoro, M.; Gay, F. Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies. Cancers 2019, 11, 2015. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef] [Green Version]
- De Veirman, K.; Menu, E.; Maes, K.; De Beule, N.; De Smedt, E.; Maes, A.; Vlummens, P.; Fostier, K.; Kassambara, A.; Moreaux, J.; et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. 2019, 442, 233–241. [Google Scholar] [CrossRef]
- Chong, P.S.; Zhou, J.; Lim, J.S.; Hee, Y.T.; Chooi, J.-Y.; Chung, T.-H.; Tan, Z.T.; Zeng, Q.; Waller, D.D.; Sebag, M.; et al. IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma. Cancer Res. 2019, 79, 4679–4688. [Google Scholar] [CrossRef]
- Lin, Y.-H.T.; Way, G.P.; Barwick, B.G.; Mariano, M.C.; Marcoulis, M.; Ferguson, I.D.; Driessen, C.; Boise, L.H.; Greene, C.S.; Wiita, A.P. Integrated phosphoproteomics and transcriptional classifiers reveal hidden RAS signaling dynamics in multiple myeloma. Blood Adv. 2019, 3, 3214–3227. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.-H.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitkopf, S.B.; Yuan, M.; Helenius, K.P.; Lyssiotis, C.A.; Asara, J.M. Triomics Analysis of Imatinib-Treated Myeloma Cells Connects Kinase Inhibition to RNA Processing and Decreased Lipid Biosynthesis. Anal. Chem. 2015, 87, 10995–11006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, H.; Du, J.; Li, L.; Li, R.; Lu, J.; Fu, W.; Hou, J. USP15 inhibits multiple myeloma cell apoptosis through activating a feedback loop with the transcription factor NF-κBp65. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Liu, C.; Ge, F.; Xiao, C.; Lu, C.; Wang, T.; He, Q.-Y. Identification of ubiquitinated proteins from human multiple myeloma U266 cells by proteomics. Biomed. Environ. Sci. 2011, 24, 422–430. [Google Scholar]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-κB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.-T.; Anderson, K.C.; Shaughnessy, J.D.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Westhrin, M.; Bondt, A.; Wuhrer, M.; Standal, T.; Holst, S. Serum protein N-glycosylation changes in multiple myeloma. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 960–970. [Google Scholar] [CrossRef]
- Natoni, A.; Farrell, M.L.; Harris, S.; Falank, C.; Kirkham-McCarthy, L.; Macauley, M.S.; Reagan, M.R.; Dwyer, M.O. Sialyltransferase inhibition leads to inhibition of tumor cell interactions with E-selectin, VCAM1, and MADCAM1, and improves survival in a human multiple myeloma mouse model. Haematologica 2019, 105, 457–467. [Google Scholar] [CrossRef]
- Harada, T.; Ohguchi, H.; Grondin, Y.; Kikuchi, S.; Sagawa, M.; Tai, Y.-T.; Mazitschek, R.; Hideshima, T.; Anderson, K.C. HDAC3 regulates DNMT1 expression in multiple myeloma: Therapeutic implications. Leukemia 2017, 31, 2670–2677. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Ma, D.; Cheng, B.; Fang, Q.; Kuang, X.; Yu, K.; Wang, W.; Hu, B.; Wang, J.; Kuang, X. Crucial role of HO-1/IRF4-dependent apoptosis induced by panobinostat and lenalidomide in multiple myeloma. Exp. Cell Res. 2018, 363, 196–207. [Google Scholar] [CrossRef]
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.-T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31, 2661–2669. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liao, R.; Yu, Y.; Zhai, H.; Wang, Y.; Sack, R.; Peters, A.H.F.M.; Chen, J.; Wu, H.; Huang, Z.; et al. Absolute Quantification of Histone PTM Marks by MRM-Based LC-MS/MS. Anal. Chem. 2014, 86, 9679–9686. [Google Scholar] [CrossRef]
- Ge, F.; Xiao, C.-L.; Yin, X.-F.; Lu, C.-H.; Zeng, H.-L.; He, Q.-Y. Phosphoproteomic analysis of primary human multiple myeloma cells. J. Proteom. 2010, 73, 1381–1390. [Google Scholar] [CrossRef]
- Brown, R.; Yang, S.; Weatherburn, C.; Gibson, J.; Ho, P.J.; Suen, H.; Hart, D.; Joshua, D. Phospho-flow detection of constitutive and cytokine-induced pSTAT3/5, pAKT and pERK expression highlights novel prognostic biomarkers for patients with multiple myeloma. Leukemia 2014, 29, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, D.S.; Das, A.; Ray, A.; Song, Y.; Samur, M.K.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Blockade of Deubiquitylating Enzyme USP1 Inhibits DNA Repair and Triggers Apoptosis in Multiple Myeloma Cells. Clin. Cancer Res. 2017, 23, 4280–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, M.; Schick, M.; Keller, U.; Krönke, J. Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers 2020, 12, 3764. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Reiman, T.; Maxwell, C.A.; Taylor, B.J.; Larratt, L.M.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; I Aronson, L.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; A Walker, B.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seval, G.C.; Beksac, M. A comparative safety review of histone deacetylase inhibitors for the treatment of myeloma. Expert Opin. Drug Saf. 2019, 18, 563–571. [Google Scholar] [CrossRef]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef]
- Xu, J.; Sun, H.-Y.; Xiao, F.-J.; Wang, H.; Yang, Y.; Wang, L.; Gao, C.-J.; Guo, Z.-K.; Wu, C.-T.; Wang, L.-S. SENP1 inhibition induces apoptosis and growth arrest of multiple myeloma cells through modulation of NF-κB signaling. Biochem. Biophys. Res. Commun. 2015, 460, 409–415. [Google Scholar] [CrossRef]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; Dinardo, C.D.; Daver, N. Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discov. 2020, 10, 506–525. [Google Scholar] [CrossRef] [Green Version]
- Takami, M.; Katayama, K.; Noguchi, K.; Sugimoto, Y. Protein kinase C alpha-mediated phosphorylation of PIM-1L promotes the survival and proliferation of acute myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2018, 503, 1364–1371. [Google Scholar] [CrossRef]
- Brown, F.C.; Still, E.; Koche, R.P.; Yim, C.Y.; Takao, S.; Cifani, P.; Reed, C.; Gunasekera, S.; Ficarro, S.B.; Romanienko, P.; et al. MEF2C Phosphorylation Is Required for Chemotherapy Resistance in Acute Myeloid Leukemia. Cancer Discov. 2018, 8, 478–497. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Aasebø, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of insulin and pathway inhibitors on the PI3K-Akt-mTOR phosphorylation profile in acute myeloid leukemia cells. Signal Transduct. Target. Ther. 2019, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Cao, M.; Wu, S.; Wang, L.; Hu, J.; Mehran, R.J.; Roth, J.A.; Swisher, S.G.; Wang, R.-Y.; Kantarjian, H.M.; et al. Anti-leukemia activity of NSC-743380 in SULT1A1-expressing acute myeloid leukemia cells is associated with inhibitions of cFLIP expression and PI3K/AKT/mTOR activities. Oncotarget 2017, 8, 102150–102160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R.; et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 2018, 131, 2698–2711. [Google Scholar] [CrossRef]
- Wang, H.; Bei, L.; Shah, C.A.; Huang, W.; Platanias, L.C.; Eklund, E.A. The E3 ubiquitin ligase Triad1 influences development of Mll-Ell-induced acute myeloid leukemia. Oncogene 2018, 37, 2532–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barghout, S.H.; Patel, P.S.; Wang, X.; Xu, G.W.; Kavanagh, S.; Halgas, O.; Zarabi, S.F.; Gronda, M.; Hurren, R.; Jeyaraju, D.V.; et al. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia 2019, 33, 37–51. [Google Scholar] [CrossRef]
- Dong, S.; Chen, J. SUMOylation of sPRDM16 promotes the progression of acute myeloid leukemia. BMC Cancer 2015, 15, 893. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef]
- Reiter, K.; Polzer, H.; Krupka, C.; Maiser, A.; Vick, B.; Rothenberg-Thurley, M.; Metzeler, K.H.; Dörfel, D.; Salih, H.R.; Jung, G.; et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 2018, 32, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, K.; Xie, Y.; Burcu, M.; Linn, D.E.; Qiu, Y.; Baer, M.R. Pim-1 Kinase Phosphorylates and Stabilizes 130 kDa FLT3 and Promotes Aberrant STAT5 Signaling in Acute Myeloid Leukemia with FLT3 Internal Tandem Duplication. PLoS ONE 2013, 8, e74653. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, X.; Cheng, Z.; Zhu, J.; Xu, L.; Wang, F.; Qi, W.; Yan, J.; Liu, N.; Sun, Z.; et al. Quantitative Analysis of Global Proteome and Lysine Acetylome Reveal the Differential Impacts of VPA and SAHA on HL60 Cells. Sci. Rep. 2016, 6, 19926. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.-J.; Lin, Y.-J.; Chao, M.-W.; Sung, T.-Y.; Wu, Y.-W.; Chen, Y.-Y.; Lin, M.-H.; Liou, J.-P.; Pan, S.-L.; Yang, C.-R. The anticancer effects of MPT0G211, a novel HDAC6 inhibitor, combined with chemotherapeutic agents in human acute leukemia cells. Clin. Epigenetics 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zhu, Y.; Lin, Y.-C.; Li, M.; Du, J.; Dong, H.; Sun, J.; Zhu, L.; Wang, H.; Ding, Z.; et al. PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2019, 134, 548–560. [Google Scholar] [CrossRef]
- Dosil, M.; Wang, S.; Lemischka, I.R. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol. Cell. Biol. 1993, 13, 6572–6585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.; Tamura, K.; DeAngelo, D.J.; De Bono, J.; Lorente, D.; Minden, M.; Uy, G.L.; Kantarjian, H.; Chen, L.S.; Gandhi, V.; et al. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br. J. Cancer 2018, 118, 1425–1433. [Google Scholar] [CrossRef]
- Nogami, A.; Okada, K.; Ishida, S.; Akiyama, H.; Umezawa, Y.; Miura, O. Inhibition of the STAT5/Pim Kinase Axis Enhances Cytotoxic Effects of Proteasome Inhibitors on FLT3-ITD–Positive AML Cells by Cooperatively Inhibiting the mTORC1/4EBP1/S6K/Mcl-1 Pathway. Transl. Oncol. 2019, 12, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Saland, E.; Vergez, F.; Serhan, N.; Delabesse, E.; De Mas, V.M.-; Hospital, M.-A.; Tamburini, J.; Manenti, S.; Sarry, J.E.; et al. Antileukemic Activity of 2-Deoxy-d-Glucose through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia with FLT3-ITD or c-KIT Mutations. Mol. Cancer Ther. 2015, 14, 2364–2373. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, M.; Paolillo, R.; Piechaczyk, M.; Bossis, G. The SUMO Pathway in Hematomalignancies and Their Response to Therapies. Int. J. Mol. Sci. 2019, 20, 3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Huang, F.-F.; Wu, D.-S.; Li, W.-J.; Zhan, H.-E.; Peng, M.-Y.; Fang, P.; Cao, P.-F.; Zhang, M.-M.; Zeng, H.; et al. SUMOylation of insulin-like growth factor 1 receptor, promotes proliferation in acute myeloid leukemia. Cancer Lett. 2015, 357, 297–306. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting histone methyltransferase and demethylase in acute myeloid leukemia therapy. OncoTargets Ther. 2017, ume 11, 131–155. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.-Y.; Yu, L. Epigenetic therapies in acute myeloid leukemia: The role of hypomethylating agents, histone deacetylase inhibitors and the combination of hypomethylating agents with histone deacetylase inhibitors. Chin. Med J. 2020, 133, 699–715. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, J.; Mead, A.J. Heterogeneity in myeloproliferative neoplasms: Causes and consequences. Adv. Biol. Regul. 2019, 71, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiology 2019, 86, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, A.U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T. The Molecular Pathology of Myelodysplastic Syndrome. Pathobiology 2018, 86, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoi, K.; Cross, N.C. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Cmoch, A.; Wolczyk, M.; Bugajski, L.; Tkaczyk, M.; Dadlez, M.; Nieborowska-Skorska, M.; Koromilas, A.E.; Skorski, T.; Piwocka, K. Increased phosphorylation of eIF2α in chronic myeloid leukemia cells stimulates secretion of matrix modifying enzymes. Oncotarget 2016, 7, 79706–79721. [Google Scholar] [CrossRef] [Green Version]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2V617Fcauses PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Xu, C.; Yang, C.; Zuo, H.; Chen, C.; Zhang, D.; Shi, G.; Wang, W.; Shi, J.; Zhang, T. Discovery and evaluation of ZT55, a novel highly-selective tyrosine kinase inhibitor of JAK2V617F against myeloproliferative neoplasms. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef]
- Gu, C.; Feng, M.; Yin, Z.; Luo, X.; Yang, J.; Li, Y.; Li, T.; Wang, R.; Fei, J. RalA, a GTPase targeted by miR-181a, promotes transformation and progression by activating the Ras-related signaling pathway in chronic myelogenous leukemia. Oncotarget 2016, 7, 20561–20573. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Dwivedi, T.R.; Yengkhom, R.; Bheemsetty, V.A.; Abe, T.; Kiyonari, H.; VijayRaghavan, K.; Inamdar, M.S. Asrij/OCIAD1 suppresses CSN5-mediated p53 degradation and maintains mouse hematopoietic stem cell quiescence. Blood 2019, 133, 2385–2400. [Google Scholar] [CrossRef]
- Park, C.S.; Lewis, A.H.; Chen, T.J.; Bridges, C.S.; Shen, Y.; Suppipat, K.; Puppi, M.; Tomolonis, J.A.; Pang, P.D.; Mistretta, T.-A.; et al. A KLF4-DYRK2–mediated pathway regulating self-renewal in CML stem cells. Blood 2019, 134, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Tvorogov, D.; Thomas, D.; Liau, N.P.D.; Dottore, M.; Barry, E.F.; Lathi, M.; Kan, W.L.; Hercus, T.R.; Stomski, F.; Hughes, T.P.; et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci. Adv. 2018, 4, eaat3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.; Leo, E.; Takemaru, K.-I.; Campi, V.; Castagnetti, F.; Soverini, S.; De Benedittis, C.; Rosti, G.; Cavo, M.; Santucci, M.A.; et al. 14-3-3 Binding and Sumoylation Concur to the Down-Modulation of β-catenin Antagonist chibby 1 in Chronic Myeloid Leukemia. PLoS ONE 2015, 10, e0131074. [Google Scholar] [CrossRef] [Green Version]
- Spiciarich, D.R.; Oh, S.T.; Foley, A.; Hughes, S.B.; Mauro, M.J.; Abdel-Wahab, O.; Press, R.D.; Viner, R.; Thompson, S.L.; Chen, Q.; et al. A Novel Germline Variant in CSF3R Reduces N-Glycosylation and Exerts Potent Oncogenic Effects in Leukemia. Cancer Res. 2018, 78, 6762–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecquet, C.; Chachoua, I.; Roy, A.; Balligand, T.; Vertenoeil, G.; Leroy, E.; Albu, R.-I.; Defour, J.-P.; Nivarthi, H.; Hug, E.; et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood 2019, 133, 2669–2681. [Google Scholar] [CrossRef]
- Sun, J.; He, X.; Zhu, Y.; Ding, Z.; Dong, H.; Feng, Y.; Du, J.; Wang, H.; Wu, X.; Zhang, L.; et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell Stem Cell 2018, 23, 355–369.e9. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Sharma, V.; Horvat, N.P.; Akuffo, A.A.; Beatty, M.S.; Murdun, C.; Colin, C.; Billington, J.M.R.; Goodheart, W.E.; Sahakian, E.; et al. HDAC11 deficiency disrupts oncogene-induced hematopoiesis in myeloproliferative neoplasms. Blood 2020, 135, 191–207. [Google Scholar] [CrossRef]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 360. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, K.; Kawazu, M.; Koya, J.; Yoshimi, A.; Masamoto, Y.; Maki, H.; Toya, T.; Kobayashi, T.; Nannya, Y.; Arai, S.; et al. A germline HLTF mutation in familial MDS induces DNA damage accumulation through impaired PCNA polyubiquitination. Leukemia 2019, 33, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Fujino, T.; Sheridan, P.; Zhang, Y.-Z.; Nagase, R.; Horikawa, S.; Li, Z.; Matsui, H.; Kanai, A.; Saika, M.; et al. A novel ASXL1– OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia 2018, 32, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. Developmental Therapeutics in Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk. 2017, 17, S43–S52. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Marcellino, B.; Lu, M.; Kremyanskaya, M.; Fabris, F.; Sandy, L.; Mehrotra, M.; Houldsworth, J.; Najfeld, V.; El Jamal, S.; et al. A phase I study of panobinostat and ruxolitinib in patients with primary myelofibrosis (PMF) and post--polycythemia vera/essential thrombocythemia myelofibrosis (post--PV/ET MF). Leuk. Res. 2020, 88, 106272. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Suo, J.; Katayama, H.; Wei, Y.; Garcia-Manero, G.; Hanash, S. Quantitative proteomic analysis of histone modifications in decitabine sensitive and resistant leukemia cell lines. Clin. Proteom. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.L.; Nairismägi, M.-L.; Laurensia, Y.; Lim, J.-Q.; Tan, J.; Li, Z.-M.; Pang, W.-L.; Kizhakeyil, A.; Wijaya, G.-C.; Huang, D.-C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2017, 24, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Murga-Zamalloa, C.; Rolland, D.C.; Polk, A.; Wolfe, A.; Dewar, H.; Chowdhury, P.; Önder, Ö.; Dewar, R.; Brown, N.A.; Bailey, N.G.; et al. Colony-Stimulating Factor 1 Receptor (CSF1R) Activates AKT/mTOR Signaling and Promotes T-Cell Lymphoma Viability. Clin. Cancer Res. 2019, 26, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C. Current targeted therapies in lymphomas. Am. J. Health Pharm. 2019, 76, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Rolland, D.; Basrur, V.; Conlon, K.; Wolfe, T.; Fermin, D.; Nesvizhskii, A.I.; Lim, M.S.; Elenitoba-Johnson, K.S. Global Phosphoproteomic Profiling Reveals Distinct Signatures in B-Cell Non-Hodgkin Lymphomas. Am. J. Pathol. 2014, 184, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Gengenbacher, A.; Müller-Rudorf, A.; Poggio, T.; Gräßel, L.; Dumit, V.I.; Kreutmair, S.; Lippert, L.J.; Duyster, J.; Illert, A.L. Proteomic Phosphosite Analysis Identified Crucial NPM-ALK-Mediated NIPA Serine and Threonine Residues. Int. J. Mol. Sci. 2019, 20, 4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valla, K.; Flowers, C.R.; Koff, J.L. Targeting the B cell receptor pathway in non-Hodgkin lymphoma. Expert Opin. Investig. Drugs 2018, 27, 513–522. [Google Scholar] [CrossRef]
- Saint-Germain, E.; Mignacca, L.; Huot, G.; Acevedo, M.; Moineau-Vallée, K.; Calabrese, V.; Bourdeau, V.; Rowell, M.-C.; Ilangumaran, S.; Lessard, F.; et al. Phosphorylation of SOCS1 Inhibits the SOCS1–p53 Tumor Suppressor Axis. Cancer Res. 2019, 79, 3306–3319. [Google Scholar] [CrossRef]
- Yang, Y.; Kelly, P.; Shaffer, A.L.; Schmitz, R.; Yoo, H.M.; Liu, X.; Huang, D.W.; Webster, D.; Young, R.M.; Nakagawa, M.; et al. Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse Large B Cell Lymphoma. Cancer Cell 2016, 29, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Ding, M.; Wang, C.; Yang, X.; Ye, T.; Yu, H. TRIM11 promotes lymphomas by activating the β-catenin signaling and Axin1 ubiquitination degradation. Exp. Cell Res. 2020, 387, 111750. [Google Scholar] [CrossRef]
- Vishwamitra, D.; Curry, C.V.; Shi, P.; Alkan, S.; Amin, H.M. SUMOylation Confers Posttranslational Stability on NPM-ALK Oncogenic Protein. Neoplasia 2015, 17, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Salahuddin, S.; Fath, E.K.; Biel, N.; Ray, A.; Moss, C.R.; Patel, A.; Patel, S.; Hilding, L.; Varn, M.; Ross, T.; et al. Epstein-Barr Virus Latent Membrane Protein-1 Induces the Expression of SUMO-1 and SUMO-2/3 in LMP1-positive Lymphomas and Cells. Sci. Rep. 2019, 9, 208. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Han, Y.; Lu, W.; Yang, J.; Tian, J.; Sun, P.; Yu, T.; Hu, Y.; Zhang, H.; et al. Overexpression of GLT1D1 induces immunosuppression through glycosylation of PD-L1 and predicts poor prognosis in B-cell lymphoma. Mol. Oncol. 2020, 14, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017, 7, 322–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaming, H.; McLean, C.M.; Korthout, T.; Alemdehy, M.F.; Hendriks, S.; Lancini, C.; Palit, S.; Klarenbeek, S.; Kwesi-Maliepaard, E.M.; Molenaar, T.M.; et al. Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L-dose dependency in HDAC1-deficient thymic lymphoma. EMBO J. 2019, 38, e101564. [Google Scholar] [CrossRef]
- Lu, X.; Fernando, T.M.; Lossos, C.; Yusufova, N.; Liu, F.; Fontán, L.; Durant, M.; Geng, H.; Melnick, J.; Luo, Y.; et al. PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood 2018, 132, 2026–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Al-Hussein, K.A.; Manogaran, P.S.; Wickrema, A.; Gutierrez, M.I.; Bhatia, K.G. Inhibition of Phosphatidylinositol 3′-Kinase/AKT Signaling Promotes Apoptosis of Primary Effusion Lymphoma Cells. Clin. Cancer Res. 2005, 11, 3102–3108. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.P.; Bhende, P.M.; Sin, S.-H.; Roy, D.; Dittmer, D.P.; Damania, B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 2010, 115, 4455–4463. [Google Scholar] [CrossRef]
- Mohanty, S.; Kumar, S.S.; Das, P.; Sahu, S.K.; Choudhuri, T. Multi-targeted therapy of everolimus in Kaposi’s sarcoma associated herpes virus infected primary effusion lymphoma. Apoptosis 2017, 22, 1098–1115. [Google Scholar] [CrossRef]
- Wong, J.P.; Stuhlmiller, T.J.; Giffin, L.C.; Lin, C.; Bigi, R.; Zhao, J.; Zhang, W.; Cruz, A.G.B.; Park, S.I.; Earp, H.S.; et al. Kinome profiling of non-Hodgkin lymphoma identifies Tyro3 as a therapeutic target in primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 2019, 116, 16541–16550. [Google Scholar] [CrossRef] [Green Version]
- Bubman, D.; Guasparri, I.; Cesarman, E. Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene 2007, 26, 4979–4986. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Staudt, L.M. Protein ubiquitination in lymphoid malignancies. Immunol. Rev. 2014, 263, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runckel, K.; Barth, M.J.; Mavis, C.; Gu, J.J.; Hernandez-Ilizaliturri, F.J. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018, 2, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Luo, Y.; Jiang, X.; Lu, X.; Roberti, D.; Lossos, C.; Kunkalla, K.; Magistri, M.; Rui, L.; Verdun, R.; et al. Recent BCR stimulation induces a negative autoregulatory loop via FBXO10 mediated degradation of HGAL. Leukemia 2019, 34, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bouchlaka, M.N.; Wolff, J.; Grindle, K.M.; Lu, L.; Qian, S.; Zhong, X.; Pflum, N.; Jobin, P.; Kahl, B.S.; et al. FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene 2016, 35, 6223–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derenzini, E.; Mondello, P.; Erazo, T.; Portelinha, A.; Liu, Y.; Scallion, M.; Asgari, Z.; Philip, J.; Hilden, P.; Valli, D.; et al. BET Inhibition-Induced GSK3β Feedback Enhances Lymphoma Vulnerability to PI3K Inhibitors. Cell Rep. 2018, 24, 2155–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delforoush, M.; Berglund, M.; Edqvist, P.; Sundström, C.; Gullbo, J.; Enblad, G. Expression of possible targets for new proteasome inhibitors in diffuse large B-cell lymphoma. Eur. J. Haematol. 2016, 98, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Sun, Y.; Wang, J.; He, Q.; Chen, X.; Lan, X.; Chen, J.; Dou, Q.P.; Shi, X.; Liu, J. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2019, 38, 453. [Google Scholar] [CrossRef]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathog. 2020, 9, 543. [Google Scholar] [CrossRef]
- Mohanty, S.; Han, T.; Choi, Y.B.; Lavorgna, A.; Zhang, J.; Harhaj, E.W. The E3/E4 ubiquitin conjugation factor UBE4B interacts with and ubiquitinates the HTLV-1 Tax oncoprotein to promote NF-κB activation. PLoS Pathog. 2020, 16, e1008504. [Google Scholar] [CrossRef]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [Green Version]
- Selby, T.L.; Biel, N.; Varn, M.; Patel, S.; Patel, A.; Hilding, L.; Ray, A.; Ross, T.; Cramblet, W.T.; Moss, C.R.; et al. The Epstein-Barr Virus Oncoprotein, LMP1, Regulates the Function of SENP2, a SUMO-protease. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Suzuki, O. Glycosylation in lymphoma: Biology and glycotherapy. Pathol. Int. 2019, 69, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, O.; Nozawa, Y.; Kawaguchi, T.; Abe, M. Alpha-2,6-sialylation of L-PHA reactive oligosaccharides and expression of N-acetylglucosaminyltransferase V in human diffuse large B cell lymphoma. Oncol. Rep. 2003, 10, 1759–1764. [Google Scholar] [CrossRef]
- Suzuki, O.; Nozawa, Y.; Abe, M. Loss of L-PHA-, PNA-, or ConA-reactive oligosaccharides is associated with a poor progno-sis in human Burkitt’s lymphoma. Oncol. Rep. 2007, 17, 775–779. [Google Scholar] [PubMed] [Green Version]
- Suzuki, O.; Abe, M.; Hashimoto, Y. Sialylation and glycosylation modulate cell adhesion and invasion to extracellular matrix in human malignant lymphoma: Dependency on integrin and the Rho GTPase family. Int. J. Oncol. 2015, 47, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, N.; Haimovich, J. Altered N-Linked Glycosylation in Follicular Lymphoma and Chronic Lymphocytic Leukemia: Involvement in Pathogenesis and Potential Therapeutic Targeting. Front. Immunol. 2017, 8, 912. [Google Scholar] [CrossRef]
- Linley, A.; Krysov, S.; Ponzoni, M.; Johnson, P.W.; Packham, G.; Stevenson, F.K. Lectin binding to surface Ig variable regions provides a universal persistent activating signal for follicular lymphoma cells. Blood 2015, 126, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Hatzi, K.; Geng, H.; Doane, A.S.; Meydan, C.; Lariviere, R.; Cardenas, M.; Duy, C.; Shen, H.; Vidal, M.N.C.; Baslan, T.; et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat. Immunol. 2019, 20, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.-M.; Huang, Y.-H.; Huang, J.-Y.; Wang, Z.-F.; Fu, D.; Liu, H.; Liu, F.; Leboeuf, C.; Wang, L.; Ye, J.; et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica 2018, 103, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Lue, J.K.; Prabhu, S.A.; Liu, Y.; Gonzalez, Y.; Verma, A.; Mundi, P.S.; Abshiru, N.; Camarillo, J.M.; Mehta, S.; Chen, E.I.; et al. Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clin. Cancer Res. 2019, 25, 5271–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]





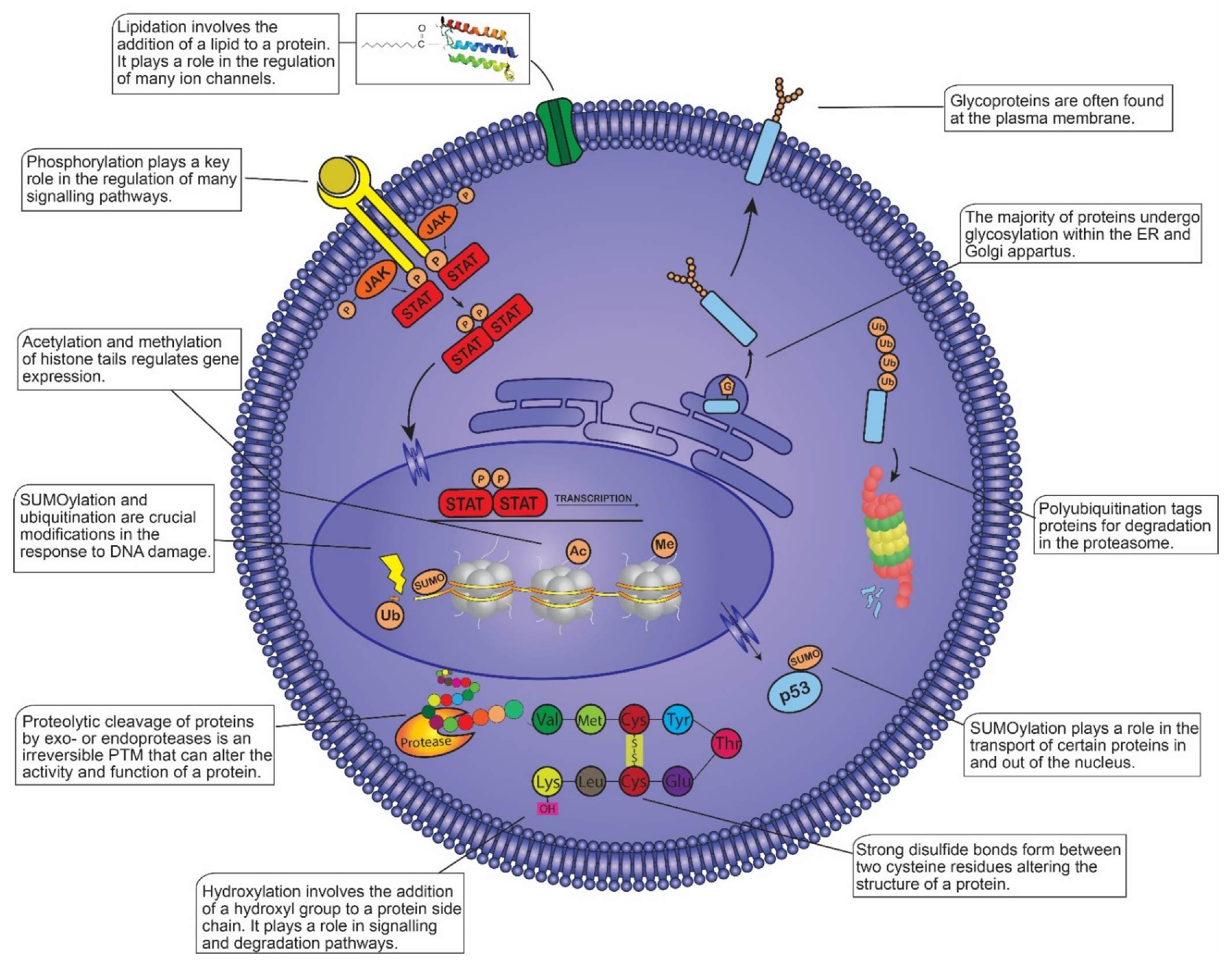{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Therapeutic | Drug | PTM Affected | Type of Blood Cancer | Mechanism of Action | References |
|---|---|---|---|---|---|
| Kinase Inhibitors (KIs) | Ruxolitinib (JAKAFI®) | Phosphorylation | Myelofibrosis Polycythemia Vera | JAK2 inhibitor | [191,192] |
| Midostaurin (RYDAPT®) | Phosphorylation | FLT3-mutant AML, Advanced systemic mastocytosis (AdvSM) | FLT3 inhibitor in AML. KIT inhibitor in AdvSM. | [193,194] | |
| Gilteritinib (XOSPATA®) | Phosphorylation | FLT3-mutant Acute myeloid leukemia (AML) | FLT3, ALK inhibitor | [195] | |
| Imatinib (GLEEVEC®) | Phosphorylation | Ph+ Chronic myeloid leukemia (CML), Ph+ Acute lymphoblastic leukemia (ALL), Myelodysplastic/ myeloproliferative diseases (MDS/MPD), Aggressive systemic mastocytosis (ASM), Chronic eosinophilic leukemia (CEL) | BCR-ABL inhibitor | [196] | |
| Dasatinib (SPRYCEL®) | Phosphorylation | Ph+ CML, Ph+ ALL | BCR-ABL, SRC inhibitor | [197] | |
| Nilotinib (TASIGNA®) | Phosphorylation | Ph+ CML | BCR-ABL inhibitor | [198] | |
| Bosutinib (BOSULIF®) | Phosphorylation | Ph+ CML | BCR-ABL and SRC inhibitor | [199] | |
| Ponatinib (ICLUSIG®) | Phosphorylation | CML, Ph+ ALL | BCR-ABL inhibitor | [200] | |
| Ibrutinib (IMBRUVICA®) | Phosphorylation | Mantle cell lymphoma (MCL), Chronic lymphocytic leukaemia (CLL) Small lymphocytic lymphoma (SLL) Waldenström’s macroglobulinemia (WM) Marginal zone lymphoma (MZL) | BTK inhibitor | [201] | |
| Idelalisib (ZYDELIG®) | Phosphorylation | CLL, SLL Follicular lymphoma (FL) | Phosphatidylinositol 3-kinase delta (PI3Kδ) inhibitor | [202] | |
| Proteasome Inhibitors (PIs) | Bortezomib (VELCADE®) | Ubiquitination | MCL, Multiple myeloma (MM) | 26S proteasome inhibitor | [203] |
| Carfilzomib (KYPROLIS®) | Ubiquitination | MM | 26S proteasome inhibitor | [204] | |
| Ixazomib (NINLARO®) | Ubiquitination | MM | 26S proteasome inhibitor | [205] | |
| Differentiation Therapy | All-trans retinoic acid (ATRA) (VESANOID®) and arsenic trioxide (TRISENOX®) | Sumoylation Ubiquitination | Acute promyelocytic leukemia (APL) | Sumoylation-dependent degradation of the fusion oncoprotein PML-RARα. | [206] |
| Immunomodulatory Drugs (IMiDs) | Lenalidomide (REVLIMID®) | Ubiquitination | MM, MDS, MCL, FL, MZL | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [207] |
| Thalidomide (THALOMID®) | Ubiquitination | MM | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [208] | |
| Pomalidomide (POMALYST®) | Ubiquitination | MM | Modulation of CRL4CRBN E3 ubiquitin ligase activity. | [208] | |
| Histone Deacetylase Inhibitors (HDACi) | Panobinostat (FARYDAK®) | Acetylation | MM | Pan-HDAC inhibitor | [209] |
| Vorinostat (ZOLINZA®) | Acetylation | Cutaneous T cell lymphoma (CTCL) | Class I, II HDAC inhibitor | [210] | |
| Belinostat (BELEODAQ®) | Acetylation | Peripheral T cell lymphoma (PTCL) | Pan-HDAC inhibitor | [211] | |
| Romidepsin (ISTODAX®) | Acetylation | CTCL, PTCL | Class I HDAC inhibitor | [212] |
| PTM Analyzed | Proteomic Technique | Main Finding | Reference |
|---|---|---|---|
| Phosphorylation | Western blot analysis | Myeloid-derived suppressor cells (MDSCs) drive enhanced phosphorylation of AMPK, promoting MM cell survival | [215] |
| Phosphorylation | Western blot analysis | PRL-3 aberrantly phosphorylates STAT3 through SHP-2 repression, leading to constant activation of STAT3 | [216] |
| Phosphorylation | Trypsin digestion, IMAC phosphopeptide enrichment, LC–MS/MS, Western blot analysis | Elucidated signaling dynamics in MM to aid precision medicine | [217] |
| Phosphorylation | Trypsin digestion, IMAC phosphopeptide enrichment, LC–MS/MS, Western blot analysis | Bone marrow stromal cells stimulate the activation, through phosphorylation, of JAK/STAT signaling. Tofacitinib reverses BMSC-mediated proliferation of MM cells | [218] |
| Phosphorylation | SILAC labeling, trypsin digestion, SCX chromatography, IMAC phosphopeptide enrichment, phosphotyrosine immunoprecipitation, LC–MS/MS | MM cells treated with imatinib show inhibition of kinase activity due to RNA processing and a decrease in lipid biosynthesis | [219] |
| Ubiquitination | Human influenza hemagglutinin (HA)-tagged ubiquitin, immunoprecipitation, Western blot analysis | Destabilization of NEK2, via its ubiquitination, reduces MM cell growth and overcomes resistance to proteasome inhibitors | [220] |
| Ubiquitination | Immunoprecipitation, immunoblot analysis | USP15 inhibits ubiquitination and degradation of NF-κBp65 which in turn promotes USP15 expression resulting in a feedback loop enhancing MM survival | [221] |
| Ubiquitination | Immunoprecipitation, SDS-PAGE, trypsin digestion, LC–MS/MS | Identification of 73 ubiquitination sites on 52 ubiquitinated proteins in human MM U266 cells | [222] |
| SUMOylation | Immunoprecipitation, Western blot analysis | Downregulation of SENP2 increases IκBα sumoylation which activates NF-κB, leading to bortezomib resistance | [223] |
| SUMOylation | Cell culture, transfection, SDS-PAGE, Western blot analysis, chemiluminescence, co-immunoprecipitation | Identification of a sumoylation signature in MM that is associated with adverse clinical outcome | [224] |
| N-glycosylation | HILIC-solid phase extraction, MALDI–TOF–MS | Analysis of serum protein N-glycosylation in MM revealed a correlation between N-glycosylation marks and ISS stages | [225] |
| Glycosylation (Sialylation) | Lectin histochemistry, lectins: Sambucus Nigra (SNA), Peanut Agglutinin (PNA), Maackia Amurensis Lectin II (MALII) | Inhibition of sialylation prevents MM cell interactions with E-selectin, MADCAM1 and VCAM1 restricting the access of tumor cells to the protective BM microenvironment | [226] |
| Acetylation | Site directed mutagenesis, SDS-PAGE, immunoblot analysis | Inhibition of HDAC3 and DNMT1 reduces survival of MM cells | [227] |
| Acetylation | SDS-PAGE, immunoblot analysis | FDA-approved panobinostat increases acetylation of H3K9 resulting in IRF4 inhibition and MM cell apoptosis | [228] |
| Methylation | SDS-PAGE, immunoblot analysis | KDM6B, independent of demethylase activity, upregulates MAPK signaling, leading to MM survival and proliferation | [229] |
| Methylation Acetylation | Isotopic labeling, -Multiple reactionmonitoring based LC–MS/MS, label-free quantification | Quantification of histone PTM marks in MM cell line | [230] |
| PTM Analyzed | Proteomic Technique | Main Finding | Reference |
|---|---|---|---|
| Phosphorylation | Immunoprecipitation, Western blot analysis | Phosphorylation of the oncogenic kinase PIM-1L by PKCα stimulates proliferation and growth of AML cells | [246] |
| Phosphorylation | Trypsin digestion, iTRAQ labeling, Fe3+-IMAC, RP-SAX-RP, LC–MS, Western blot analysis | Resistance to chemotherapy in AML requires the phosphorylation of transcription factor, MEF2C | [247] |
| Phosphorylation | SILAC, filter-aided sample preparation (FASP), LC–MS | Analysis of the impact of insulin and specific inhibitors on the phosphorylation of PI3K-Akt-mTOR pathway components in AML cells | [248] |
| Phosphorylation | Western blot analysis | Gilteritinib reduces phosphorylation of FLT3 and its downstream targets, improving the survival of FLT3-mutated AML mouse models | [249] |
| Phosphorylation | Reverse-phase protein microarray (RPPA), Western blot analysis | STAT3 inhibitor, NSC-743380, induces apoptosis in SULT1A1-expressing AML cells by inhibiting the activity of the PI3K/AKT/mTOR pathway | [250] |
| Ubiquitination | Site-directed mutagenesis, immunoprecipitation, Western blot analysis | Ubiquitination and degradation of CDK2 results in the differentiation of AML cells through the activation of PRDX2 | [251] |
| Ubiquitination Phosphorylation | Western blot analysis | E3 ubiquitin ligase, TRIAD1, suppresses leukemogenesis in 11q23-AML | [252] |
| Ubiquitination | Immunoblot analysis, immunohistochemistry | Inhibitor of E1 ubiquitin-activating enzyme, Uba1, TAK-243 selectively decreases growth and survival of AML cells. | [253] |
| SUMOylation | Site-directed mutagenesis, immunoprecipitation, Western blot analysis | SUMOylation of sPRDM16, promotes AML progression and inhibits differentiation | [254] |
| SUMOylation | Western blot analyses | SUMOylation inhibitor, 2-D08, inhibits AML cell viability through ROS accumulation-mediated apoptosis | [255] |
| Phosphorylation Glycosylation | Western blot analysis | Tyrosine kinase inhibition increases surface expression of FLT3 via increased glycosylation, demonstrating therapeutic potential in combination with FLT3-directed therapy | [256] |
| Phosphorylation Ubiquitination Glycosylation | Immunoprecipitation, Western blot analysis | Serine/threonine kinase PIM-1 stabilizes unglycosylated FLT3-ITD, promoting the activation of STAT5 signaling | [257] |
| Acetylation | SILAC labeling, HPLC fractionation, immunoaffinity enrichment, LC–MS/MS | HDAC inhibitors have differential impacts on the lysine acetylome in AML cells | [258] |
| Acetylation Phosphorylation | Immunoprecipitation, Western blot analysis | Combination of novel HDAC inhibitor, MPT0G211, with current chemotherapeutics has anti-proliferative effects on human AML cells | [259] |
| Methylation | Mutagenesis, Western blot analysis | Methylation of FLT3-ITD by PRMT1 supports the persistence of FLT3-ITD+ AML cells | [260] |
| PTM Analyzed | Proteomic Technique | Main Finding | Reference |
|---|---|---|---|
| Phosphorylation | SILAC, FASP, in-gel isoelectric focusing, HPLC fractionation, LC–MS/MS, Western blot analysis | Enhanced phosphorylation of eukaryotic initiation factor 2 α subunit (eIF2α) increases the secretion of extracellular enzymes, promoting the invasiveness of CML cells | [277] |
| Phosphorylation | Western blot analysis | MPNs with JAK2V617F mutation evade the immune system through upregulated PD-L1 expression | [278] |
| Phosphorylation | Western blot analysis | Selective inhibitor of mutant JAK2 (V617F), ZT55, inhibits proliferation and induces apoptosis in HEL cell line | [279] |
| Phosphorylation | Western blot analysis, phospho-specific protein microarray analysis | Increased RalA, a small GTPase, promotes malignant transformation and progression in CML through the activation of Ras signaling. | [280] |
| Ubiquitination | Immunoblot analysis | The stem cell protein, Asrij/OCIAD1, prevents the degradation of p53, thus mediating hematopoietic stem cell quiescence and preventing uncontrolled proliferation | [281] |
| Phosphorylation Ubiquitination | Immunoprecipitation, immunoblot analysis | KLF4 promotes leukemogenesis through the repression of DYRK2, which mediates the activation of p53 and the degradation of c-Myc in CML-like disease in mice | [282] |
| Phosphorylation Ubiquitination | Immunoprecipitation, Western blot analysis, TUBE-based ubiquitin-binding assay | The loss of LZTR1 function, a mediator of Ras ubiquitination and MAPK signaling contributes to TKI resistance in BCR-ABL CML cells. | [283] |
| Phosphorylation Ubiquitination | Immunoprecipitation, Western blot analysis, TUBE-based ubiquitin-binding assay | Type I JAK inhibitor, but not a type II inhibitor, mediates pathogenic withdrawal signaling through the accumulation of phosphorylated JAK2 by preventing dephosphorylation and ubiquitination. | [284] |
| Phosphorylation SUMOylation | Immunoprecipitation, Western blot analysis | Down-modulation of the β-catenin antagonist, CBY1, in CML is induced by 14-3-3 binding and enhanced SUMOylation followed by proteasomal degradation | [285] |
| Phosphorylation Glycosylation | Immunoprecipitation, Western blot analysis, TMT labeling, N-glycan permethylation, MALDI–TOF–MS | Mutations in CSF3R prevents N-glycosylation, promoting ligand-independent activation of the JAK/STAT pathway | [286] |
| Phosphorylation Glycosylation | Immunoprecipitation, Western blot analysis, size-exclusion chromatography, MS-based analysis | Mutant calreticulin (CALR) acts as a rogue chaperone thrombopoietin receptor (TpoR), immature TpoR and mutant TpoR resulting in cytokine-independent activation and constituent JAK/STAT activation | [287] |
| Acetylation | Western blot analysis, immunoprecipitation, SDS-PAGE, in-gel digestion, LC–MS/MS | Activation of deacetylase Sirtuin 1 (SIRT1) restores Tet methylcytosine dioxygenase 2 (TET2) activity, disrupting the maintenance of MDS HSPCs. | [288] |
| Acetylation | Immunoprecipitation, Western blot analysis, LC–MS/MS | Inhibition of HDAC11 downregulates the JAK/STAT pathway and induces apoptosis in MPLW515L-MPN mouse model | [289] |
| Phosphorylation Methylation | Immunoprecipitation, Western blot analysis | Overexpression of the histone demethylase, JMJD1C, in MPNs prevents H3K9me2 and HP1α-mediated repression of NFE2 resulting in a positive feedback loop | [290] |
| PTM Analyzed | Proteomic Technique | Main Finding | Reference |
|---|---|---|---|
| Phosphorylation | Immunohistochemistry, Western blot analysis | Increased STAT3 phosphorylation promotes PD-L1 expression in NKTL | [300] |
| Phosphorylation | Western blot analysis, PhosphoFlow cytometry, reverse phase protein microarray, TiO2 enrichment, LC–MS/MS | The dual PI3K/mTOR inhibitor, PQR309, demonstrates anti-cancer activity alone and in combination with other therapies in preclinical lymphoma models | [301] |
| Phosphorylation | Immunohistochemistry, Western blot analysis, LC–MS/MS | Phosphorylation of SOCS1 by Src family kinases inhibits SOCS1-p53-mediated senescence | [308] |
| Phosphorylation | Trypsin digestion, TiO2 enrichment, tyrosine-phosphorylated peptide immunoprecipitation, LC–MS/MS, Western blot analysis | CSF1R expression is altered in many T cell lymphomas. CSF1R activation by CSF1 leads to phosphorylation and activation of downstream PI3K/AKT/mTOR signaling, thus promoting proliferation and survival | [302] |
| Ubiquitination | Site-directed mutagenesis, immunoprecipitation, Western blot analysis, TUBE assay | The ubiquitin ligases cIAP1 and cIAP2 promote oncogenic BCR signaling and represent the potential of SMAC mimetics for the treatment of ABC DLBCL | [309] |
| Ubiquitination | Western blot analysis | Enhanced expression of TRIM11 activates β-catenin signaling via Axin1 ubiquitination and degradation, thus promoting lymphomagenesis | [310] |
| SUMOylation | Immunoprecipitation, Western blot analysis, in vitro SUMOylation assay, site-directed mutagenesis | Dysregulated SUMOylation in NPM-ALK+ T-cell lymphoma stabilizes the NPM-ALK fusion protein resulting in its accumulation promoting carcinogenesis | [311] |
| SUMOylation | Western blot analysis | Epstein-Barr Virus LMP1 enhances SUMOylation through interaction with the SUMO E2-conjugating enzyme, Ubc9, in LMP1-positive lymphomas | [312] |
| Glycosylation | Western blot analysis, glycoprotein deglycosylation assay, GC-MS | Overexpression of the glycosyltransferase, GLT1D1, is a poor prognostic marker that enhances PD-L1 glycosylation promoting tumor growth in B cell NHL | [313] |
| Acetylation | Western blot analysis | The tumor suppressing acetyltransferase, CREBBP, is haploinsufficient in GC-derived B cell NHL | [314] |
| Acetylation Methylation Ubiquitination | Western blot analysis, nanoLC-MRM, MS/MS | HDAC1-deficient thymic lymphomas show increased H3K79 methylation and demonstrate sensitivity to a DOT1L inhibitor | [315] |
| Methylation | Immunoprecipitation, Western blot analysis, in vitro methyltransferase assay, SDS-PAGE, trypsin digestion, LC–MS/MS | PRMT5-mediated regulation of BCL6, via methylation, is required for germinal center formation. Dual inhibition of PRMT5 and BCL6 suppresses DLBCL proliferation | [316] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunphy, K.; Dowling, P.; Bazou, D.; O’Gorman, P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers 2021, 13, 1930. https://doi.org/10.3390/cancers13081930
Dunphy K, Dowling P, Bazou D, O’Gorman P. Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers. Cancers. 2021; 13(8):1930. https://doi.org/10.3390/cancers13081930
Chicago/Turabian StyleDunphy, Katie, Paul Dowling, Despina Bazou, and Peter O’Gorman. 2021. "Current Methods of Post-Translational Modification Analysis and Their Applications in Blood Cancers" Cancers 13, no. 8: 1930. https://doi.org/10.3390/cancers13081930