
Current Perspectives on the Use of off the Shelf CAR-T/NK Cells for the Treatment of Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
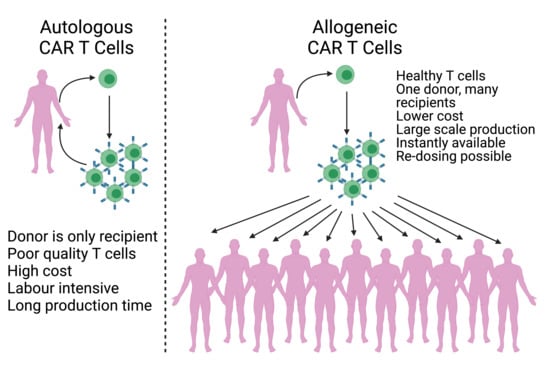
2. Autologous and Allogeneic CAR T Cells
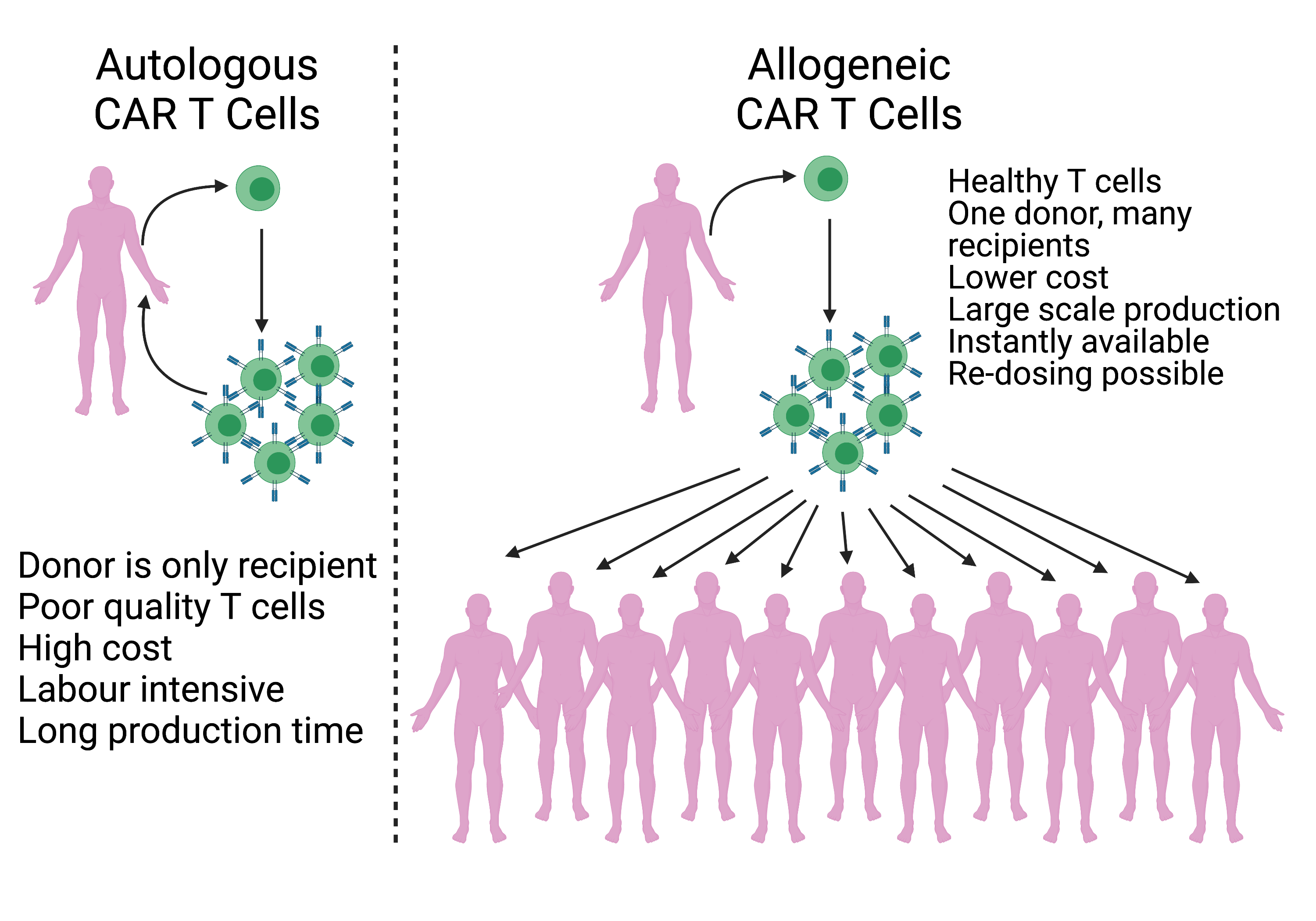
3. γδ. T Cells
4. NK Cells
5. Invariant NK T Cells
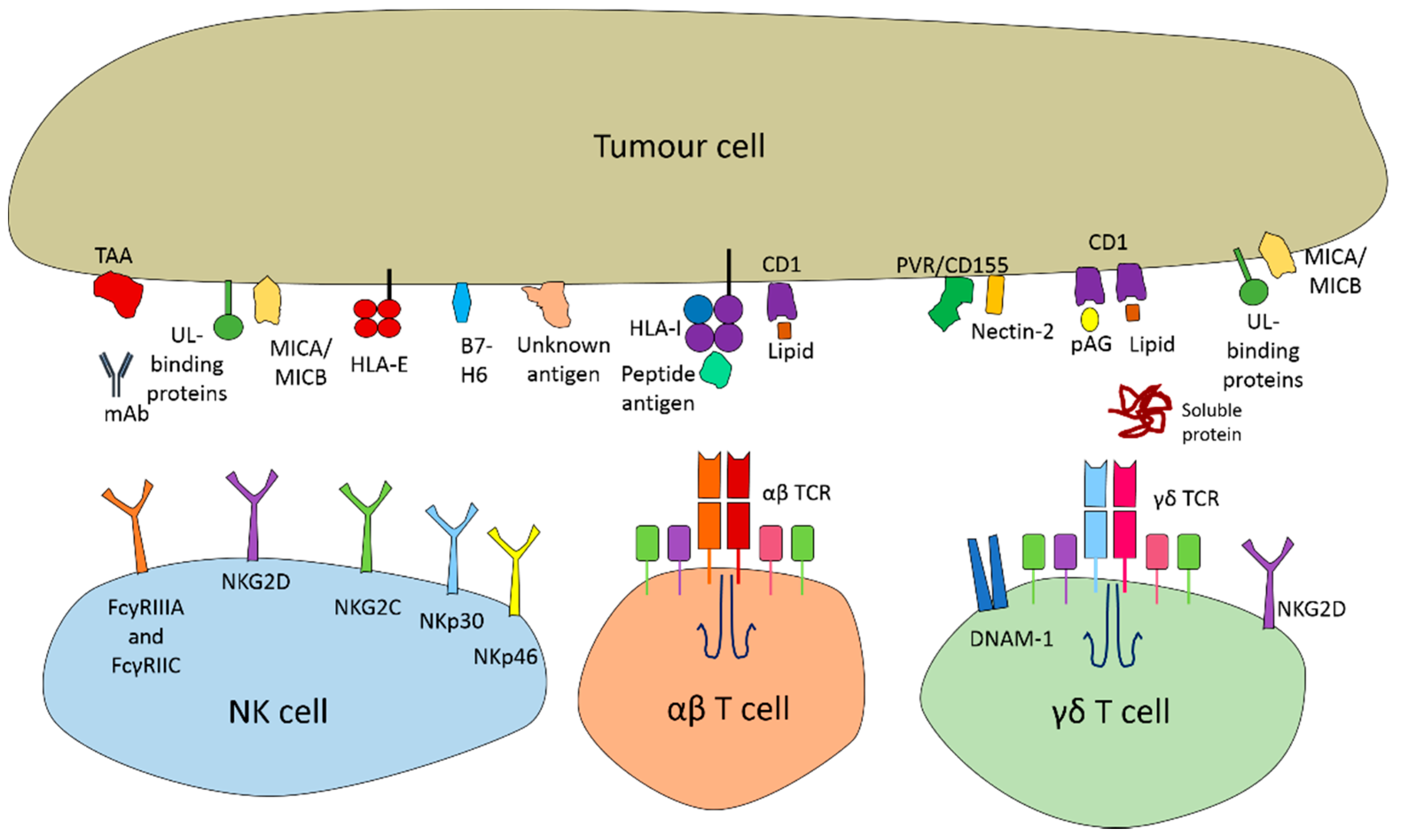
6. Genome Editing of αβ T Cells
7. Induced Pluripotent Stem Cells
8. Conclusions
Funding
Conflicts of Interest
References
- Burnet, M. Cancer—A biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: Fromtumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The immune system in cancer pathogenesis: Potential therapeutic approaches. J. Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of resistance to immune checkpoint blockade: Why does checkpoint inhibitor immunotherapy not work for all patients? Am. Soc. Clin. Oncol. Educ. Book. 2019, 1, 147–164. [Google Scholar] [CrossRef]
- Wang, X.; Waschke, B.C.; Woolaver, R.A.; Chen, Z.; Zhang, G.; Piscopio, A.D.; Liu, X.; Wang, J.H. Histone deacetylase inhibition sensitizes pd1 blockade–resistant B-cell lymphomas. Cancer Immunol Res. 2019, 7, 1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance mechanisms to immune—checkpoint blockade in cancer: Tumor-intrinsic and -extrinsic factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967. [Google Scholar] [CrossRef]
- Bioley, G.; Guillaume, P.; Luescher, I.; Yeh, A.; Dupont, B.; Bhardwaj, N.; Mears, G.; Old, L.J.; Valmori, D.; Ayyoub, M. HLA class I–associated immunodominance affects CTL Responsiveness to an ESO recombinant protein tumor antigen vaccine. Clin. Cancer Res. 2009, 15, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.-Y.; Wu, S.-P.; Liao, R.-Q.; Huang, S.-M.; Wu, Y.-L. Potential biomarker for checkpoint blockade immunotherapy and treatment strategy. Tumor Biol. 2016, 37, 4251–4261. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer. 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T.; et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and design of chimeric antigen receptors. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Petrovic, R.M.G.; Kao, Y.V.; Saxena, S.A.; Romain, A.; Costa-Guerra, J.A.; Violette, S.; et al. Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol. Ther. 2017, 25, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration, CfDEaR. YESCARTA (axicabtagene ciloleucel), STN:BL 125643, 31 March 2017. Available online: https://www.fda.gov/media/108458/download (accessed on 26 February 2021).
- U.S. Food and Drug Administration, CfDEaR. TECARTUS (brexucabtagene autoleucel), STN:BL 125703, 24 July 2020. Available online: https://www.fda.gov/media/140415/download (accessed on 26 February 2021).
- U.S. Food and Drug Administration, CfDEaR. KYMRIAH (tisagenlecleucel), STN:BL 125646, 30 August 2017. Available online: https://www.fda.gov/media/106989/download (accessed on 26 February 2021).
- Herper, M.H. Novartis CEO’s Dilemma: Is $475,000 Too Much for a Leukaemia Breakthrough? Or Is It No Enough? 2017. Available online: https://www.forbes.com/sites/matthewherper/2017/08/30/novartis-ceos-dilemma-is-475000-too-much-for-a-leukemia-breakthrough-or-is-it-not-enough/?sh=1e9d21a556ec (accessed on 26 February 2021).
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA approval summary: Tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res. 2019, 25, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.V.; Porter, D.L. Graft-versus-host disease after donor leukocyte infusions: Presentation and management. Clin. Haematol. 2008, 21, 205–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Kalyan, S.; Kabelitz, D. Defining the nature of human γδ T cells: A biographical sketch of the highly empathetic. Cell Mol. Immunol. 2013, 10, 21–29. [Google Scholar] [CrossRef]
- Kabelitz, D.; Wesch, D.; He, W. Perspectives of γδ T cells in tumor immunology. Cancer Res. 2007, 67, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Hayday, A.C. γδ Cells: A right time and a right place for a conserved third way of protection. Annu. Rev. Immunol. 2000, 18, 975–1026. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Holtmeier, W.; Pfänder, M.; Hennemann, A.; Zollner, T.M.; Kaufmann, R.; Caspary, W.F. The TCR-delta repertoire in normal human skin is restricted and distinct from the TCR-delta repertoire in the peripheral blood. J. Investig. Derm. 2001, 116, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Kabelitz, D.; Kalyan, S.; Oberg, H.H.; Wesch, D. Human Vδ2 versus non-Vδ2 γδ T cells in antitumor immunity. Oncoimmunology 2013, 2, e23304. [Google Scholar] [CrossRef]
- Porcelli, S.A.; Modlin, R.L. The CD1 system: Antigen-presenting molecules for t cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 1999, 17, 297–329. [Google Scholar] [CrossRef]
- Russano, A.M.; Bassotti, G.; Agea, E.; Bistoni, O.; Mazzocchi, A.; Morelli, A.; Porcelli, S.A.; Spinozzi, F. CD1-restricted recognition of exogenous and self-lipid antigens by duodenal gammadelta+ T lymphocytes. J. Immunol. 2007, 178, 3620–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Libero, G.; Mori, L. Recognition of lipid antigens by T cells. Nat. Rev. Immunol. 2005, 5, 485–496. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Viey, E.; Fromont, G.; Escudier, B.; Morel, Y.; Da Rocha, S.; Chouaib, S.; Caignard, A. Phosphostim-activated γδ t cells kill autologous metastatic renal cell carcinoma. J. Immunol. 2005, 174, 1338–1347. [Google Scholar] [CrossRef]
- Morita, C.T.; Beckman, E.M.; Bukowski, J.F.; Tanaka, Y.; Band, H.; Bloom, B.R.; Golan, D.E.; Brenner, M.B. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity 1995, 3, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Shafi, S.; Vantourout, P.; Wallace, G.; Antoun, A.; Vaughan, R.; Stanford, M.; Hayday, A. An NKG2D-mediated human lymphoid stress surveillance response with high interindividual variation. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Adams, E.J.; Chien, Y.H.; Garcia, K.C. Structure of a gammadelta T cell receptor in complex with the nonclassical MHC T22. Science 2005, 308, 227–231. [Google Scholar] [CrossRef]
- Gertner-Dardenne, J.; Castellano, R.; Mamessier, E.; Garbit, S.; Kochbati, E.; Etienne, A.; Charbonnier, A.; Collette, Y.; Vey, N.; Olive, D. Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts. J. Immunol. 2012, 188, 4701–4708. [Google Scholar] [CrossRef]
- Knight, A.; Mackinnon, S.; Lowdell, M.W. Human Vdelta1 gamma-delta T cells exert potent specific cytotoxicity against primary multiple myeloma cells. Cytotherapy 2012, 14, 1110–1118. [Google Scholar] [CrossRef]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Born, W.K.; Aydintug, M.K.; O’Brien, R.L. Diversity of γδ T-cell antigens. Cell. Mol. Immunol. 2013, 10, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Raverdeau, M.; Cunningham, S.P.; Harmon, C.; Lynch, L. γδ T cells in cancer: A small population of lymphocytes with big implications. Clin. Transl. Immunol. 2019, 8, e01080. [Google Scholar] [CrossRef]
- Maniar, A.; Zhang, X.; Lin, W.; Gastman, B.R.; Pauza, C.D.; Strome, S.E.; Chapoval, A.I. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood 2010, 116, 1726–1733. [Google Scholar] [CrossRef]
- Deniger, D.C.; Maiti, S.N.; Mi, T.; Switzer, K.C.; Ramachandran, V.; Hurton, L.V.; Ang, S.; Olivares, S.; Rabinovich, B.A.; Huls, M.H.; et al. Activating and propagating polyclonal gamma delta t cells with broad specificity for malignancies. Clin. Cancer Res. 2014, 20, 5708–5719. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. NK cell receptors. Annu. Rev. Immunol. 1998, 16, 359–393. [Google Scholar] [CrossRef]
- Long, E.O. Negative signaling by inhibitory receptors: The NK cell paradigm. Immunol. Rev. 2008, 224, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valés-Gómez, M.; Reyburn, H.T.; Mandelboim, M.; Strominger, J.L. Kinetics of interaction of HLA-C ligands with natural killer cell inhibitory receptors. Immunity 1998, 9, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Uhrberg, M.; Valiante, N.M.; Shum, B.P.; Shilling, H.G.; Lienert-Weidenbach, K.; Corliss, B.; Tyan, D.; Lanier, L.L.; Parham, P. Human diversity in killer cell inhibitory receptor genes. Immunity 1997, 7, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Morel, P.A.; Ernst, L.K.; Metes, D. Functional CD32 molecules on human NK cells. Leuk. Lymphoma 1999, 35, 47–56. [Google Scholar] [CrossRef]
- Lanier, L.L.; Ruitenberg, J.J.; Phillips, J.H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 1988, 141, 3478–3485. [Google Scholar]
- Anderson, P.; Caligiuri, M.; O’Brien, C.; Manley, T.; Ritz, J.; Schlossman, S.F. Fc gamma receptor type III (CD16) is included in the zeta NK receptor complex expressed by human natural killer cells. Proc. Natl. Acad. Sci. USA 1990, 87, 2274–2278. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Re, F.; Staudacher, C.; Zamai, L.; Vecchio, V.; Bregni, M. Killer cell Ig-like receptors ligand-mismatched, alloreactive natural killer cells lyse primary solid tumors. Cancer 2006, 107, 640–648. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Klingemann, H. Challenges of cancer therapy with natural killer cells. Cytotherapy 2015, 17, 245–249. [Google Scholar] [CrossRef]
- Guven, H.; Konstantinidis, K.V.; Alici, E.; Aints, A.; Abedi-Valugerdi, M.; Christensson, B.; Ljunggren, H.-G.; Dilber, M.S. Efficient gene transfer into primary human natural killer cells by retroviral transduction. Exp. Hematol. 2005, 33, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Becker, S.; Esser, R.; Schwabe, D.; Seifried, E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J. Hematotherapy Stem Cell Res. 2001, 10, 535–544. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pietà, A.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-engineered NK-92 cells: An off-the-shelf cell therapy for prostate cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Máthé, D.; Hegedüs, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Grote, S.; Mittelstaet, J.; Baden, C.; Chan, K.C.; Seitz, C.; Schlegel, P.; Kaiser, A.; Handgretinger, R.; Schleicher, S. Adapter chimeric antigen receptor (AdCAR)-engineered NK-92 cells: An off-the-shelf cellular therapeutic for universal tumor targeting. Oncoimmunology 2020, 9, 1825177. [Google Scholar] [CrossRef]
- Nowakowska, P.; Romanski, A.; Miller, N.; Odendahl, M.; Bonig, H.; Zhang, C.; Seifried, E.; Wels, W.S.; Tonn, T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol. Immunother. 2018, 67, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, M.; Garcia, J.; Wilson, S.B.; Spada, F.; Gerdes, D.; Tahir, S.M.; Patton, K.T.; Blumberg, R.S.; Porcelli, S.; Chott, A.; et al. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology 2000, 100, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Patterson, S.; Szydlo, R.; Chaudhry, M.S.; Dazzi, F.; Kanfer, E.; McDonald, D.; Marin, D.; Milojkovic, D.; Pavlu, J.; et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood 2012, 119, 5030–5036. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Lewis, D.; Dejbakhsh-Jones, S.; Lan, F.; García-Ojeda, M.; Sibley, R.; Strober, S. Bone marrow NK1.1(-) and NK1.1(+) T cells reciprocally regulate acute graft versus host disease. J. Exp. Med. 1999, 189, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Leveson-Gower, D.B.; Olson, J.A.; Sega, E.I.; Luong, R.H.; Baker, J.; Zeiser, R.; Negrin, R.S. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood 2011, 117, 3220–3229. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell 2018, 34, 596–610.e11. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long terminal repeat CRISPR-CAR-coupled “Universal” T cells mediate potent anti-leukemic effects. Mol. Ther. 2018, 26, 1215–1227. [Google Scholar] [CrossRef] [Green Version]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Gautron, A.-S.; Juillerat, A.; Guyot, V.; Filhol, J.-M.; Dessez, E.; Duclert, A.; Duchateau, P.; Poirot, L. Fine and Predictable Tuning of TALEN gene editing targeting for improved T cell adoptive immunotherapy. Mol. Ther. Nucleic Acids 2017, 9, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Gilham, D.E.; Michaux, A.; Breman, E.; Mauen, S.; Bolsée, J.; Huberty, F.; Marijsse, J.; Violle, B.; Jacques-Hespel, C.; Marchand, C.; et al. TCR inhibitory molecule as a promising allogeneic NKG2D CAR-t cell approach. J. Clin. Oncol. 2018, 36, e15042-e. [Google Scholar] [CrossRef]
- Juillerat, A.; Tkach, D.; Yang, M.; Boyne, A.; Valton, J.; Poirot, L.; Duchateau, P. Straightforward Generation of Ultrapure Off-the-Shelf Allogeneic CAR-T Cells. Front. Bioeng. Biotechnol. 2020, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Guo, T.; Yeung, B.; Saso, K.; Anczurowski, M.; Wang, C.-H.; Murata, K.; Sugata, K.; Saijo, H.; Matsunaga, Y.; et al. Genetic ablation of HLA class I, Class II, and the T-cell receptor enables allogeneic T Cells to be used for adoptive T-cell therapy. Cancer Immunol. Res. 2020, 8, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Sheen, J.H.; Lim, O.; Lee, Y.; Ryu, J.; Shin, D.; Kim, Y.Y.; Kim, M. Abrogation of HLA surface expression using CRISPR/Cas9 genome editing: A step toward universal T cell therapy. Sci. Rep. 2020, 10, 17753. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef] [Green Version]
- Ishido, S.; Wang, C.; Lee, B.-S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300. [Google Scholar] [CrossRef]
- Wang, X.; Cabrera, F.G.; Sharp, K.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Engineering tolerance toward allogeneic CAR-T Cells by Regulation of MHC Surface Expression with Human Herpes Virus-8 Proteins. Mol. Ther. 2021, 29, 718–733. [Google Scholar] [CrossRef]
- Holling, T.M.; Van Der Stoep, N.; Quinten, E.; Elsen, P.J.V.D. Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J. Immunol. 2002, 168, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, C.; Cheng, H.-Y.; Yeung, Y.A.; Nguyen, D.; Sutton, J.; Melton, Z.; Valton, J.; Poulsen, K.; Djuretic, I.; Van Blarcom, T.; et al. Preclinical evaluation of ALLO-819, an allogeneic CAR t cell therapy targeting FLT3 for the treatment of acute myeloid leukemia. Blood 2019, 134, 3921. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, H.; Shen, X.; Du, J.; Zhang, X.; Zhao, Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017, 66, 571–578. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Valton, J.; Galetto, R.; Bentley, T.; Sutton, J.; Ni, Y.; Leonard, M.; Van Blarcom, T.; Smith, J.; et al. ALLO-715, an Allogeneic BCMA CAR T Therapy Possessing an Off-Switch for the Treatment of Multiple Myeloma. Blood 2018, 132, 591. [Google Scholar] [CrossRef]
- Cai, T.; Galetto, R.; Gouble, A.; Smith, J.; Cavazos, A.; Han, L.; Zhang, Q.; Kuruvilla, V.M.; Konoplev, S.N.; Neelapu, S.S.; et al. Pre-clinical studies of allogeneic anti-CD123 CAR T-cells for the therapy of blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood 2017, 130, 2625. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.-I.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013, 12, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Clarke, R.; Van Der Stegen, S.; Chang, C.-W.; Lai, Y.-S.; Witty, A.; Husain, M.; Wu, C.-J.; Yang, B.-H.; Dufaud, C.; et al. Abstract 3245: FT819 path to IND: First-of-kind off-the-shelf CAR19 T-cell for B cell malignancies. Cancer Res. 2020, 80, 3245. [Google Scholar]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutmore, L.; Brown, N.; Raj, D.; Chauduri, S.; Wang, P.; Maher, J.; Wang, Y.; Lemoine, N.; Marshall, J. Pancreatic Cancer UK Grand Challenge: Developments and challenges for effective CAR T cell therapy for pancreatic ductal adenocarcinoma. Pancreatology 2020, 20, 394–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
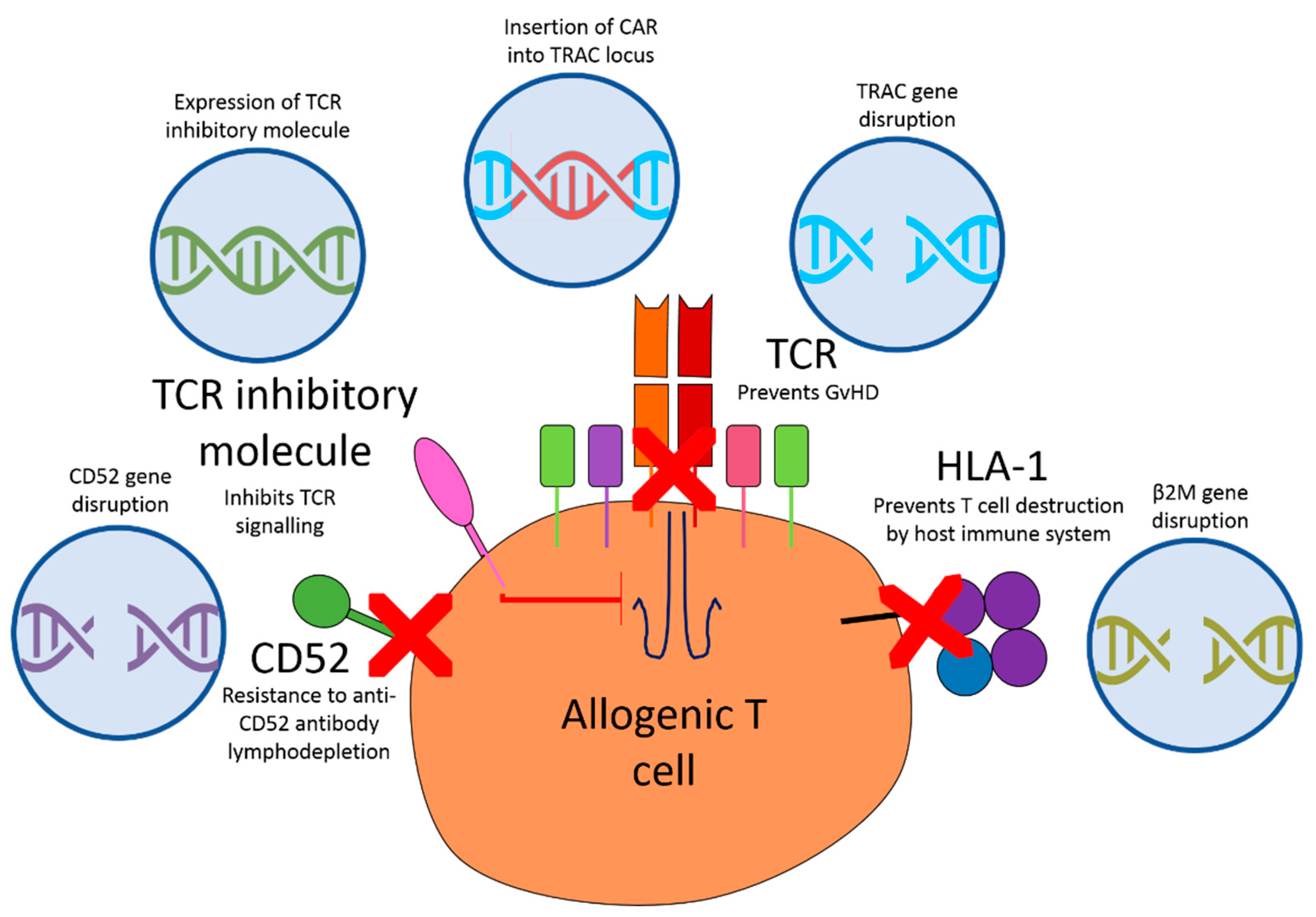{kind=link}
| Cell Type | CAR Target | Modification | Disease | Trial Number | Gene Editing Technology |
|---|---|---|---|---|---|
| γδ T cells | CD19 | Leukaemia, Lymphoma | NCT02656147 | NS | |
| γδ T cells | NKG2D ligands | Colorectal Cancer, Triple Negative Breast Cancer, Sarcoma, Nasopharyngeal Carcinoma, Prostate Cancer, Gastric Cancer | NCT04107142 | NS | |
| T cells | CD7 | TRAC disrupted | T cell Leukaemia, T cell Lymphoma | NCT04264078 | NS |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | B-cell Acute Lymphoblastic Leukaemia (B-ALL) | NCT04166838 | NS |
| T cells | CD19 | Unknown | B-cell Acute Lymphoblastic Leukaemia, B-cell Lymphoma | NCT04264039 | NS |
| T cells | CD7 | Unknown | T Acute Lymphoblastic Leukaemia and Lymphoma | NCT04620655 | NS |
| T cells | CD7 | Unknown | Haematologic Malignancies | NCT04538599 | NS |
| T cells | CD123 | Unknown | Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) | NCT03203369 | TALENs |
| T cells | BCMA | Unknown | Relapsed/Refractory Multiple Myeloma | NCT04601935 | NS |
| T cells | CD19 | Epstein-Barr Virus Specific Cytotoxic T-Lymphocytes (EBV-CTLs) | Acute Lymphocytic Leukaemia, Lymphoma | NCT01430390 | NS |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Advanced Lymphoid Malignancies | NCT02735083 | NS |
| T cells | CD19 | Unknown | Leukaemia | NCT02799550 | NS |
| T cells | CD19 | TRAC disrupted | Lymphoma | NCT04637763 | CRISPR-Cas9 |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Relapsed/Refractory Large B Cell Lymphoma | NCT04416984 | CRISPR-Cas9 |
| T cells | CD19 | TRAC disrupted | Non-Hodgkin Lymphoma, B-cell Acute Lymphoblastic Leukaemia | NCT03666000 | NS |
| T cells | BCMA | Unknown | Multiple Myeloma | NCT03752541 | CRISPR-Cas9 |
| T cells | CD123 | TRAC and CD52 genes have been disrupted | Acute Myeloid Leukaemia | NCT04106076 | TALENs |
| T cells | CD123 | TRAC and CD52 genes have been disrupted | Relapsed/Refractory Acute Myeloid Leukaemia | NCT03190278 | TALENs |
| T cells | CD123 | TRAC and CD52 genes have been disrupted | Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) | NCT03203369 | TALENs |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Adult Patients With Relapsed/Refractory B-cell Acute Lymphoblastic Leukaemia | NCT02746952 | TALENs |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Advanced Lymphoid Malignancies | NCT02735083 | TALENs |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Paediatric Relapsed/Refractory B-cell Acute Lymphoblastic Leukaemia | NCT02808442 | TALENs |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Acute Lymphoblastic Leukaemia (ALL), Non-Hodgkin Lymphoma (NHL) | NCT03229876 | CRISPR-Cas9 |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | B Cell Leukaemia, B Cell Lymphoma | NCT03166878 | CRISPR-Cas9 |
| T cells | CD19 | TRAC and CD52 genes have been disrupted | Relapsed/Refractory Lymphoma | NCT03939026 | TALENs |
| T cells | BCMA | TRAC and CD52 genes have been disrupted | Relapsed/Refractory Multiple Myeloma | NCT04093596 | TALENs |
| T cells | CD19 | MHC-1 Knockout by B2M disruption and CAR inserted into TRAC locus | B-cell Malignancy, Non-Hodgkin Lymphoma, B-cell Lymphoma | NCT04035434 | CRISPR-Cas9 |
| T cells | BCMA | MHC-1 Knockout by B2M disruption and CAR inserted into TRAC locus | Multiple Myeloma | NCT04244656 | CRISPR-Cas9 |
| T cells | CD19 | Unknown | Acute Lymphoblastic Leukaemia | NCT04154709 | CRISPR-Cas9 |
| T cells | CD22 | TRAC disrupted | B-cell Acute Lymphoblastic Leukaemia | NCT04150497 | TALENs |
| T cells | CD19 | Unknown | Diffuse Large B-cell Lymphoma | NCT04026100 | CRISPR-Cas9 |
| T cells | CD19 + CD20 or CD19+CD22 | Unknown | B Cell Leukaemia, B Cell Lymphoma | NCT03398967 | CRISPR-Cas9 |
| T cells | CS1 | TRAC disrupted | Relapsed/Refractory Multiple Myeloma | NCT04142619 | TALENs |
| T cells | CD19 | Unknown | B-cell Acute Lymphoblastic Leukaemia, B-cell Lymphoma | NCT04264039 | NS |
| T cells | CD30 | Epstein-Barr Virus-Specific T Lymphocytes | Lymphoma | NCT04288726 | NS |
| T cells | CD22 | Unknown | B Cell Malignancy | NCT04601181 | NS |
| T cells | CD19 | Unknown | B Cell Malignancy | NCT04384393 | NS |
| T cells | Unknown | Unknown | Ovarian Cancer | NCT00019136 | NS |
| T cells | CD70 | B2M KO and CAR insertion into TRAC locus | T Cell Lymphoma | NCT04502446 | CRISPR-Cas9 |
| T cells | CD19 | Unknown | ALL, Childhood B-Cell | NCT04173988 | NS |
| T cells | GD2 | Tri-virus specific cytotoxic T cells | Neuroblastoma | NCT01460901 | NS |
| T cells | CD20 | CAR inserted into TCR locus | Non-Hodgkin’s Lymphoma, | NCT04030195 | NS |
| T cells | CD19 | TRAC locus disrupted | B Acute Lymphoblastic Leukaemia | NCT04557436 | CRISPR-Cas9 |
| T cells | Cd19 | Unknown | B-cell Acute Lymphoblastic Leukaemia, B-cell Non Hodgkin Lymphoma | NCT04544592 | NS |
| T cells | CD20 | Unknown | Diffuse Large B Cell Lymphoma, Follicular Lymphoma, Mantle Cell Lymphoma, Small Lymphocytic Lymphoma | NCT04176913 | NS |
| T cells | CD19 | Unknown | B cell Lymphoma | NCT02992834 | NS |
| T cells | CD19 | Donor-derived EBV-specific cytotoxic T cells | Acute Lymphoblastic Leukaemia | NCT01195480 | NS |
| T cells | CD70 | MHC-1 Knockout by B2M disruption and CAR inserted into TRAC locus | Renal Cell Carcinoma | NCT04438083 | CRISPR-Cas9 |
| T cells | NKG2D ligands | Colorectal Cancer | NCT03692429 | Non-gene edited peptide-based technology | |
| NKT cells | CD19 | Lymphoma | NCT03774654 | NS | |
| NK cells | CD19 | NK-92 cells | Leukemia | NCT02892695 | NS |
| NK cells | CD7 | pNK cells | Leukemia | NCT02742727 | NS |
| NK cells | CD19 | CB-NK | CD19 Positive Lymphoma | NCT03579927 | NS |
| NK cells | ErbB2/Her2 | NK-92 cells | Glioblastoma | NCT03383978 | NS |
| NK cells | Unknown | NK-92 with Chimeric Costimulatory Converting Receptor (CCCR) | Non-small Cell Lung Cancer | NCT03656705 | NS |
| NK cells | CD33 | NK-92 | Leukaemia | NCT02944162 | NS |
| NK cells | BCMA | NK-92 | Multiple Myeloma | NCT03940833 | NS |
| NK cells | CD19 | NK-92 | Leukaemia, Lymphoma | NCT02892695 | NS |
| NK cells | NKG2D ligands | Solid Tumours | NCT03415100 | NS | |
| NK cells | NKG2D ligands | Membrane IL-15 | Relapsed/Refractory AML | NCT04623944 | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cutmore, L.C.; Marshall, J.F. Current Perspectives on the Use of off the Shelf CAR-T/NK Cells for the Treatment of Cancer. Cancers 2021, 13, 1926. https://doi.org/10.3390/cancers13081926
Cutmore LC, Marshall JF. Current Perspectives on the Use of off the Shelf CAR-T/NK Cells for the Treatment of Cancer. Cancers. 2021; 13(8):1926. https://doi.org/10.3390/cancers13081926
Chicago/Turabian StyleCutmore, Lauren C., and John F. Marshall. 2021. "Current Perspectives on the Use of off the Shelf CAR-T/NK Cells for the Treatment of Cancer" Cancers 13, no. 8: 1926. https://doi.org/10.3390/cancers13081926