
Using Exome Sequencing to Improve Prediction of FOLFIRINOX First Efficacy for Pancreatic Adenocarcinoma


Abstract
:Simple Summary
Abstract
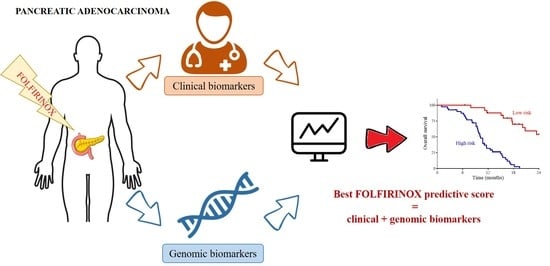
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Selection
2.3. DNA Isolation
2.4. Whole Exome Capture and Sequencing
2.5. Exome Analysis Pipeline
2.6. Statistical Analysis
3. Results
3.1. Patients’ Clinical Characteristics
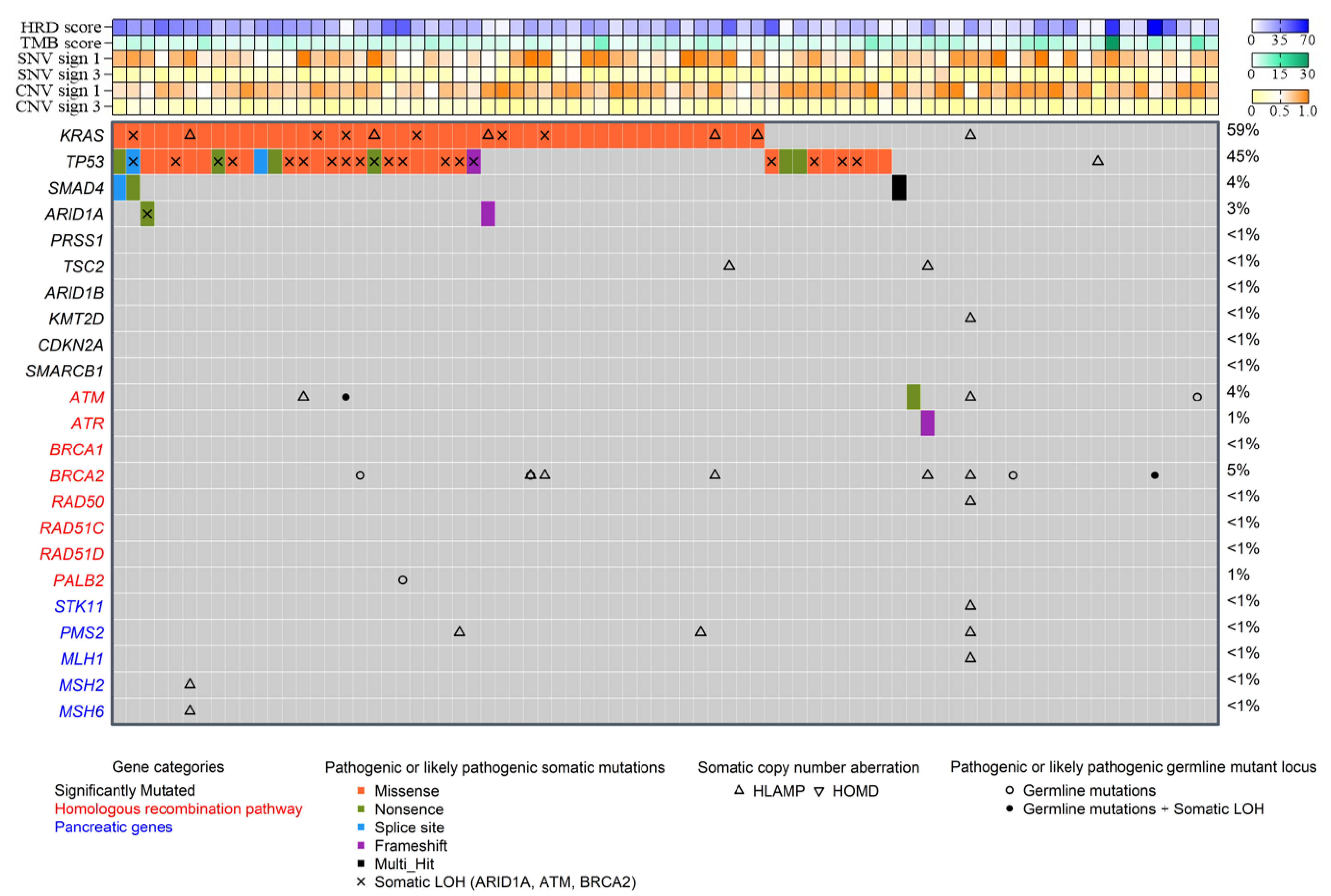
3.2. Patients’ Genomic Characteristics:
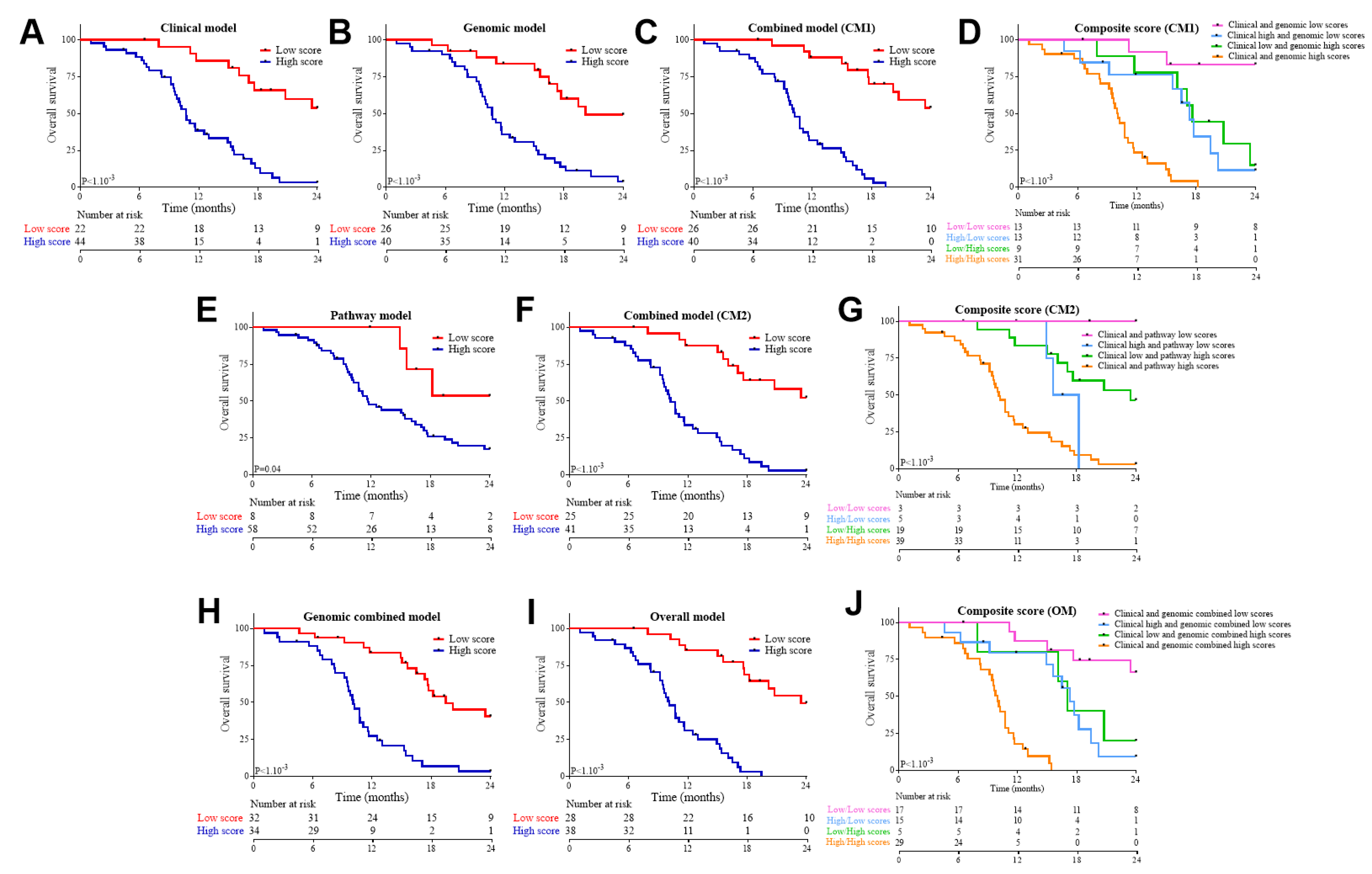
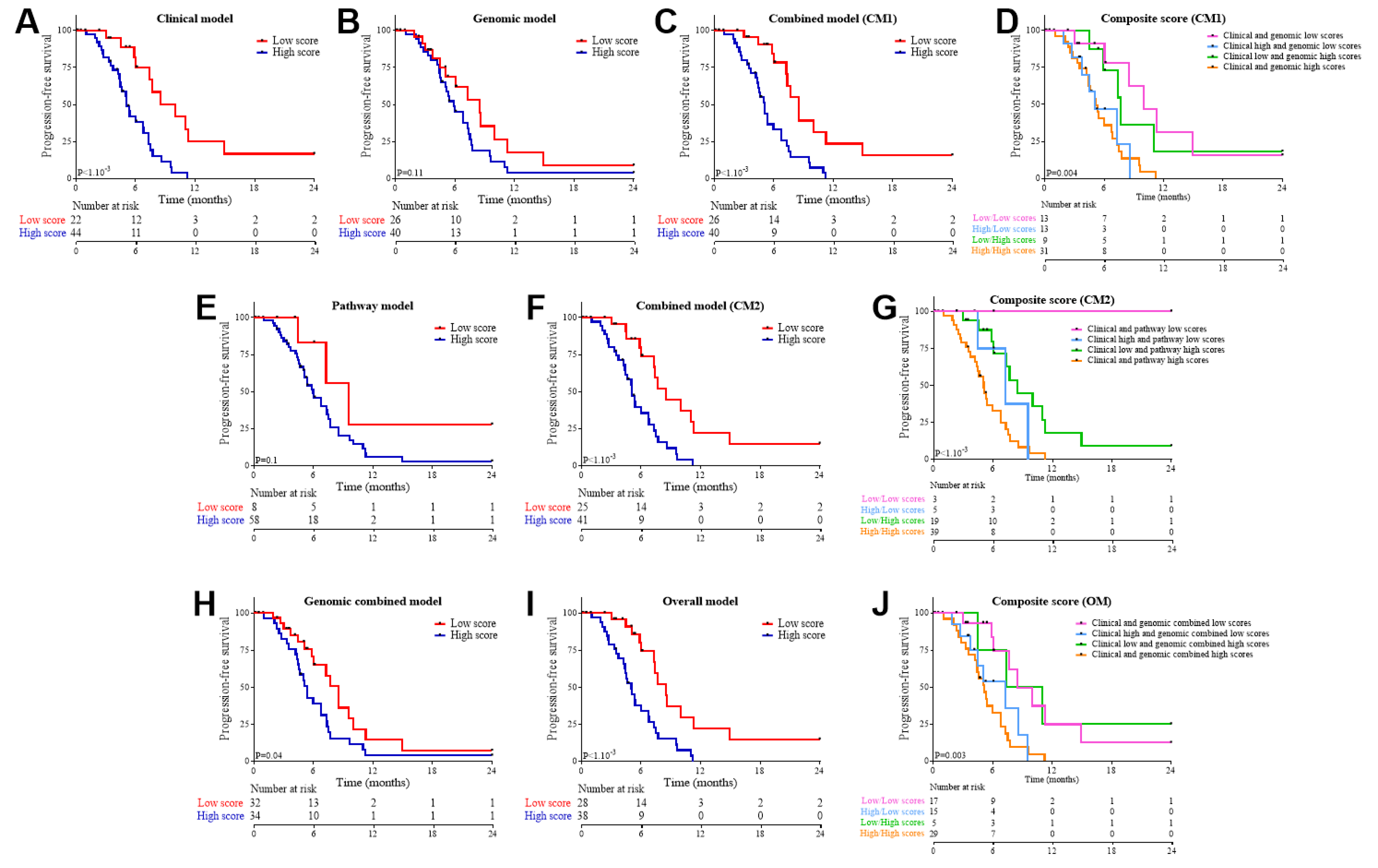
3.3. Association of Clinical and Genomic Variables with Response to FOLFIRINOX
3.4. Association of Clinical and Genomic Variables with Response to Gemcitabine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Cancer Mortality Predictions for the Year 2021 with Focus on Pancreatic and Female Lung Cancer—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0923753421000144?via%3Dihub (accessed on 18 March 2021).
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 2139–2141. [CrossRef] [PubMed] [Green Version]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, D.D.V.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-Time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From State-of-the-Art Treatments to Novel Therapies for Advanced-Stage Pancreatic Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A Phase 1 Study of LOXO-292, a Potent and Highly Selective RET Inhibitor, in Patients with RET-Altered Cancers. JCO 2018, 36, 102. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. PVAC-Seq: A Genome-Guided in Silico Approach to Identifying Tumor Neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Choe, G.; Freeman, D.J.; Castellarin, M.; Munro, S.; Moore, R.; Holt, R.A. Derivation of HLA Types from Shotgun Sequence Datasets. Genome Med. 2012, 4, 95. [Google Scholar] [CrossRef] [Green Version]
- Ha, G.; Roth, A.; Khattra, J.; Ho, J.; Yap, D.; Prentice, L.M.; Melnyk, N.; McPherson, A.; Bashashati, A.; Laks, E.; et al. TITAN: Inference of Copy Number Architectures in Clonal Cell Populations from Tumor Whole-Genome Sequence Data. Genome Res. 2014, 24, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating Mutational Processes in Single Tumors Distinguishes DNA Repair Deficiencies and Patterns of Carcinoma Evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy Number Signatures and Mutational Processes in Ovarian Carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Middha, S.; Zhang, L.; Nafa, K.; Jayakumaran, G.; Wong, D.; Kim, H.R.; Sadowska, J.; Berger, M.F.; Delair, D.F.; Shia, J.; et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP Array-Based Homologous Recombination Deficiency Measures to next Generation Sequencing Data of Breast Cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- Hothorn, T.; Lausen, B. On the Exact Distribution of Maximally Selected Rank Statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.S. Two Decades beyond BRCA1/2: Homologous Recombination, Hereditary Cancer Risk and a Target for Ovarian Cancer Therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Humphris, J.L.; Patch, A.-M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation In Pancreatic Cancer. Gastroenterology 2017, 152, 68–74.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774–783. [Google Scholar] [CrossRef]
- Sehdev, A.; Gbolahan, O.; Hancock, B.A.; Stanley, M.; Shahda, S.; Wan, J.; Wu, H.H.; Radovich, M.; O’Neil, B.H. Germline and Somatic DNA Damage Repair Gene Mutations and Overall Survival in Metastatic Pancreatic Adenocarcinoma Patients Treated with FOLFIRINOX. Clin. Cancer Res. 2018, 24, 6204–6211. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The Influence of Subclonal Resistance Mutations on Targeted Cancer Therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome Chaos: Creating New Genomic Information Essential for Cancer Macroevolution. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Whole Population (n = 78) | FOLFIRINOX Group (n = 66) | Gemcitabine Group (n = 12) | p-Value | |
|---|---|---|---|---|---|
| Clinical characteristics | Sex, n (%) | 0.53 | |||
| Men | 41 (52.6%) | 36 (54.5%) | 5 (41.7%) | ||
| Women | 37 (47.4%) | 30 (45.5%) | 7 (58.3%) | ||
| Who performance status, n (%) | 0.35 | ||||
| 0 | 30 (38.5%) | 27 (40.9%) | 3 (25%) | ||
| 1 and 2 | 48 (61.5%) | 39 (59.1%) | 9 (75%) | ||
| Stage, n (%) | 0.51 | ||||
| III | 25 (32.1%) | 20 (30.3%) | 5 (41.7%) | ||
| IV | 53 (67.9%) | 46 (69.7%) | 7 (58.3%) | ||
| Lung metastasis, n (%) | 0.28 | ||||
| No | 60 (76.9%) | 49 (74.2%) | 11 (91.7%) | ||
| Yes | 18 (23.1%) | 17 (25.8%) | 1 (8.3%) | ||
| Liver metastasis, n (%) | 0.13 | ||||
| No | 36 (46.2%) | 33 (50%) | 3 (25%) | ||
| Yes | 42 (53.8%) | 33 (50%) | 9 (75%) | ||
| Peritoneal metastasis, n (%) | 0.72 | ||||
| No | 57 (73.1%) | 49 (74.2%) | 8 (66.7%) | ||
| Yes | 21 (26.9%) | 17 (25.8%) | 4 (33.3%) | ||
| Lymph node metastasis, n (%) | 0.06 | ||||
| No | 59 (77.6%) | 47 (73.4%) | 12 (100%) | ||
| Yes | 19 (22.4%) | 17 (26.6%) | 0 | ||
| Missing data | 2 | 2 | 0 | ||
| Age at diagnosis, median (IQR) | 67.41 (13.82) | 67.41 (11.7) | 68.41 (22.78) | 0.83 | |
| Age at diagnosis, n (%) | 0.5 | ||||
| ≤60 years | 24 (30.8%) | 19 (28.2%) | 5 (41.7%) | ||
| >60 years | 54 (69.2%) | 47 (71.2%) | 7 (58.3%) | ||
| Response rate, n (%) | 0.02 | ||||
| Partial or complete response | 22 (28.2%) | 22 (33.3%) | 0 | ||
| Stabilization or progression | 48 (61.5%) | 37 (56.1%) | 11 (91.7%) | ||
| Missing data | 8 (10.3%) | 7 (10.6%) | 1 (8.3%) | ||
| OS, median (IQR) | 14.93 (11.14) | 14.93 (11.14) | 14.13 (14.9) | ||
| PFS, median (IQR) | 5.97 (4.4) | 6.77 (4.14) | 4.77 (9.17) | ||
| Genomic CharacteristCs | MSI categories, n (%) | ||||
| MSS (score ≤ 5) | 77 (98.7%) | 65 (98.5%) | 12 (100%) | 1 | |
| MSS low and high | 1 (1.3%) | 1 (1.5%) | 0 | ||
| TMB without splicing regions categories, n (%) | |||||
| Low (score ≤ 40) | 75 (96.2%) | 63 (95.5%) | 12 (100%) | 1 | |
| High | 3 (3.8%) | 3 (4.5%) | 0 | ||
| MSI score, median (IQR) | 0.04 (0.14) | 0.04 (0.13) | 0.02 (0.25) | 1 | |
| TMB without splicing regions score, median (IQR) | 3.3 (2.01) | 3.2 (1.98) | 3.8 (2.02) | 0.11 | |
| HRD score, median (IQR) | 21 (17) | 21 (18.5) | 21 (13.75) | 0.71 | |
| Ploidy, median (IQR) | 2 (0.2) | 2 (0.27) | 2 (0.22) | 0.43 | |
| Clonality, median (IQR) | 1 (1) | 1 (1) | 1 (1) | 0.74 | |
| TCR clones, median (IQR) | 5 (4) | 5 (3) | 7.5 (8.25) | 0.23 | |
| Deletions with LOH, median (IQR) | 4 (14.75) | 4 (14.75) | 5.5 (13.5) | 0.72 | |
| Microdeletions, median (IQR) | 3 (6) | 3 (6) | 2.5 (5.25) | 0.87 | |
| Neoantigens, median (IQR) | 5 (5) | 5 (5) | 5 (3) | 0.48 | |
| Strong neoantigens, median (IQR) | 1 (1) | 1 (1) | 0.5 (1.25) | 0.96 | |
| Overall Survival | Progression-Free Survival | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Variables | HR | 95% CI | p-Value | Adjusted p-Value | HR | 95% CI | p-Value | Adjusted p-Value | |
| Clinical | WHO performance status | 2.59 | [1.4; 4.86] | 0.003 | 0.02 | 2.28 | [1.16; 4.5] | 0.02 | 0.06 |
| Stage | 3.3 | [1.58; 6.88] | 0.001 | 0.01 | 3.77 | [1.75; 8.13] | <1.10−3 | 0.007 | |
| Lung Metastasis | 0.38 | [0.18; 0.79] | 0.01 | 0.02 | 0.87 | [0.44; 1.73] | 0.69 | 0.69 | |
| Liver Metastasis | 2.51 | [1.38; 4.55] | 0.002 | 0.01 | 1.82 | [0.97; 3.41] | 0.06 | 0.12 | |
| Genomic | Ploidy > 2.4 | 3.2 | [1.59; 6.47] | 0.001 | 0.007 | 1.77 | [0.84; 3.72] | 0.14 | 0,44 |
| Clonality > 1 | 0.34 | [0.17; 0.65] | 0.001 | 0.007 | 0.48 | [0.24; 0.95] | 0.03 | 0,38 | |
| CNV signature 1 >86.6% | 0.47 | [0.24; 0.93] | 0.03 | 0.09 | 0.79 | [0.39; 1.63] | 0.53 | 0.77 | |
| CNV signature 5 >4.7% | 2.62 | [1.46; 4.7] | 0.001 | 0.007 | 0.96 | [0.52; 1.78] | 0.9 | 0.9 | |
| Pathways | Calcium | 0.39 | [0.14; 1.09] | 0.07 | 0.42 | 0.48 | [0.17; 1.31] | 0.15 | 0.54 |
| Class I MHC | 2.86 | [0.86; 9.51] | 0.09 | 0.42 | 1.11 | [0.15; 8.24] | 0.92 | 0.93 | |
| MAPK | 4.87 | [1.62; 14.6] | 0.005 | 0.07 | 4.58 | [1.5; 13.9] | 0.007 | 0.15 | |
| NHEJ | 3.25 | [0.99; 10.67] | 0.05 | 0.41 | 0.45 | [0.06; 3.31] | 0.43 | 0.76 | |
| p53 | 1.61 | [0.9; 2.87] | 0.1 | 0.45 | 0.9 | [0.5; 1.58] | 0.62 | 0.87 | |
| Spliceosome | 6.28 | [2.31; 17.13] | 0.0003 | 0.009 | 3.19 | [1.1; 9.3] | 0.03 | 0.24 | |
| VEGF | 2.41 | [0.93; 6.22] | 0.07 | 0.41 | 3.7 | [1.36; 10.06] | 0.01 | 0.15 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lecuelle, J.; Aarnink, A.; Tharin, Z.; Truntzer, C.; Ghiringhelli, F. Using Exome Sequencing to Improve Prediction of FOLFIRINOX First Efficacy for Pancreatic Adenocarcinoma. Cancers 2021, 13, 1851. https://doi.org/10.3390/cancers13081851
Lecuelle J, Aarnink A, Tharin Z, Truntzer C, Ghiringhelli F. Using Exome Sequencing to Improve Prediction of FOLFIRINOX First Efficacy for Pancreatic Adenocarcinoma. Cancers. 2021; 13(8):1851. https://doi.org/10.3390/cancers13081851
Chicago/Turabian StyleLecuelle, Julie, Anne Aarnink, Zoé Tharin, Caroline Truntzer, and François Ghiringhelli. 2021. "Using Exome Sequencing to Improve Prediction of FOLFIRINOX First Efficacy for Pancreatic Adenocarcinoma" Cancers 13, no. 8: 1851. https://doi.org/10.3390/cancers13081851