
The Influence of Anti-Diabetic Drugs on Prostate Cancer

 , ,
, , {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
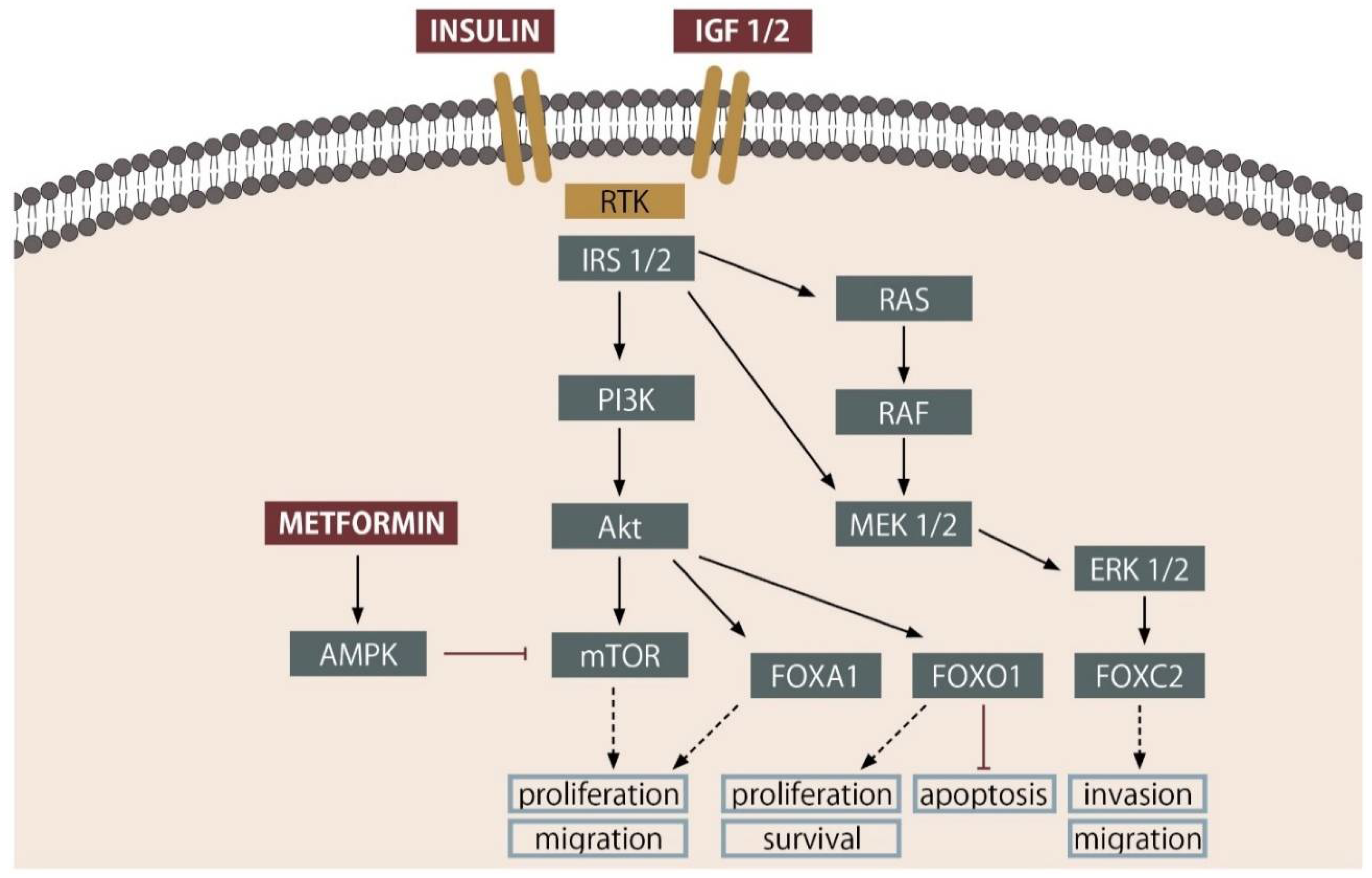
2. Insulin
3. Sulfonyloureas
4. Metformin
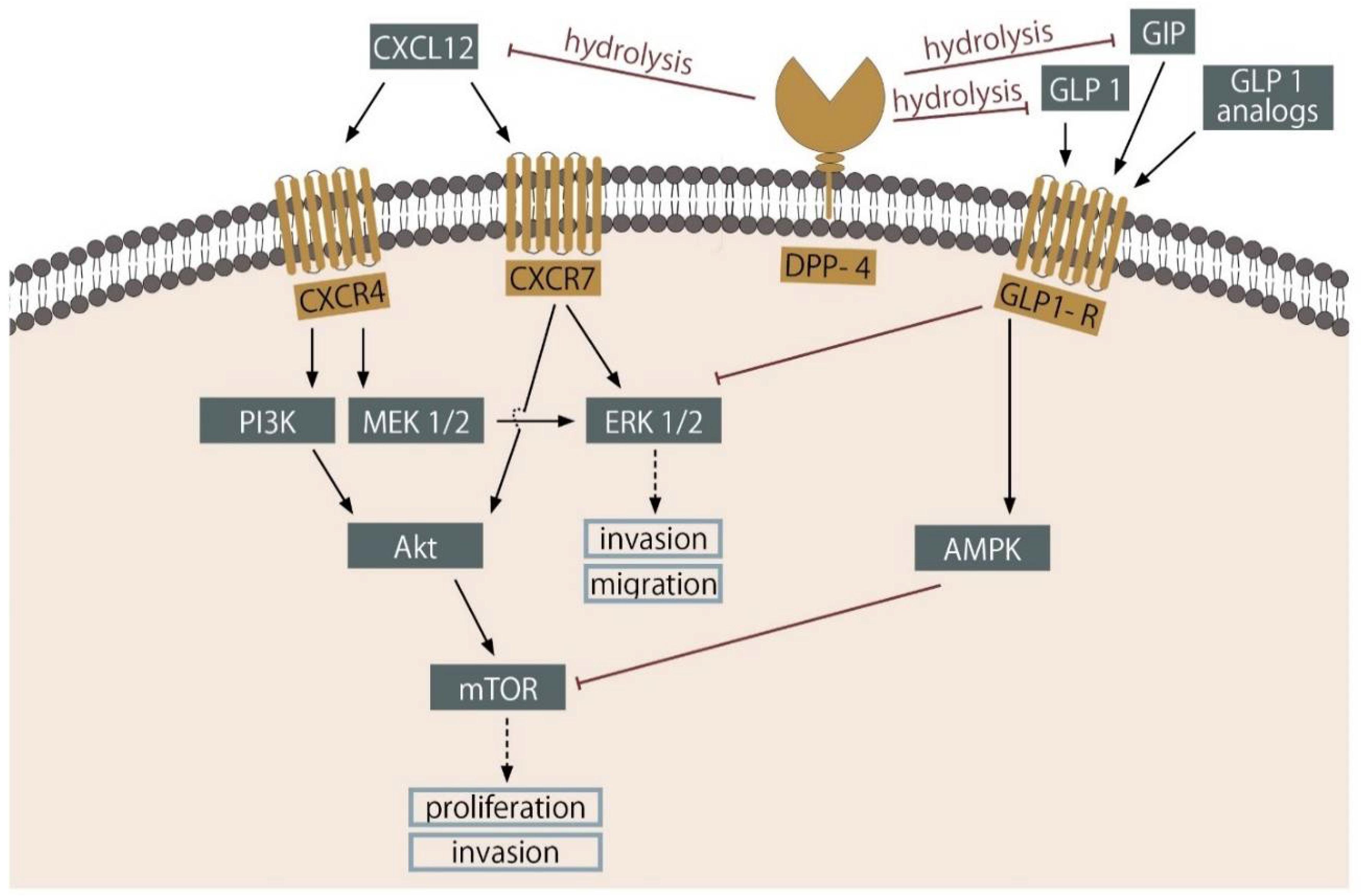
5. DPP-4 Inhibitors
6. Incretins
7. SGLT2 Inhibitors
8. Thiazolidinediones
9. Conclusions and Further Perspectives
- stratification of the risk of PC related to the treatment of T2DM,
- optimization of T2DM treatment among patients with PC (concerning the metabolic effect of ADT),
- possible use of antidiabetic drugs in the management of PC in non-diabetic patients.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Pollock, P.A.; Ludgate, A.; Wassersug, R.J. In 2124, half of all men can count on developing prostate cancer. Curr. Oncol. 2015, 22, 10–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleyer, A.; Spreafico, F.; Barr, R. Prostate cancer in young men: An emerging young adult and older adolescent challenge. Cancer 2020, 126, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.M.; Mucci, L.A. Diet and Lifestyle in Prostate Cancer. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1210, pp. 1–27. [Google Scholar]
- Mottet, N.; Van Den Bergh, R.C.N.; Vice-chair, P.C.; De Santis, M.; Gillessen, S.; Govorov, A.; Grummet, J.; Henry, A.M.; Lam, T.B.; Mason, M.D.; et al. EAU-ESUR-ESTRO-SIOG Guidelines on Prostate Cancer/Guias Europeas. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Zelenko, Z.; Gallagher, E.J. Diabetes and Cancer. Endocrinol. Metab. Clin. 2014, 43, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Zhang, P.-H.; Chen, Z.-W.; Lv, D.; Xu, Y.-Y.; Gu, W.-L.; Zhang, X.-H.; Le, Y.-L.; Zhu, H.; Zhu, Y.-M. Increased risk of cancer in patients with type 2 diabetes mellitus: A retrospective cohort study in China. BMC Public Health 2012, 12, 567. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Yang, Y.; Skrip, L.; Hu, D.; Wang, Y.; Wong, C.; Qiu, J.; Lei, H. Diabetes mellitus and risk of prostate cancer: An updated meta-analysis based on 12 case–control and 25 cohort studies. Acta Diabetol. 2012, 49, 235–246. [Google Scholar] [CrossRef]
- Bansal, D.; Bhansali, A.; Kapil, G.; Undela, K.; Tiwari, P. Type 2 diabetes and risk of prostate cancer: A meta-analysis of observational studies. Prostate Cancer Prostatic Dis. 2013, 16, 151–158. [Google Scholar] [CrossRef]
- Feng, X.; Song, M.; Preston, M.A.; Ma, W.; Hu, Y.; Pernar, C.H.; Stopsack, K.H.; Ebot, E.M.; Fu, B.C.; Zhang, Y.; et al. The association of diabetes with risk of prostate cancer defined by clinical and molecular features. Br. J. Cancer 2020, 123, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Ranc, K.; Jørgensen, M.E.; Friis, S.; Carstensen, B. Mortality after cancer among patients with diabetes mellitus: Effect of diabetes duration and treatment. Diabetologia 2014, 57, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au Yeung, S.L.; Schooling, C.M. Impact of glycemic traits, type 2 diabetes and metformin use on breast and prostate cancer risk: A Mendelian randomization study. BMJ Open Diabetes Res. Care 2019, 7, e000872. [Google Scholar] [CrossRef] [Green Version]
- Shlomai, G.; Neel, B.; LeRoith, D.; Gallagher, E.J. Type 2 Diabetes Mellitus and Cancer: The Role of Pharmacotherapy. J. Clin. Oncol. 2016, 34, 4261–4269. [Google Scholar] [CrossRef] [Green Version]
- Makrilakis, K. The Role of DPP-4 Inhibitors in the Treatment Algorithm of Type 2 Diabetes Mellitus: When to Select, What to Expect. Int. J. Environ. Res. Public Health 2019, 16, 2720. [Google Scholar] [CrossRef] [Green Version]
- IDF Diabetes Atlas. 2020. Available online: https://www.idf.org/e-library/epidemiology-research/diabetes-atlas/159-idf-diabetes-atlas-ninth-edition-2019.html (accessed on 25 April 2020).
- Care, D.; Suppl, S.S. Glycemic targets: Standards of medical care in diabetes. Diabetes Care 2020, 43, S66–S76. [Google Scholar] [CrossRef]
- Carls, G.; Huynh, J.; Tuttle, E.; Yee, J.; Edelman, S.V. Achievement of Glycated Hemoglobin Goals in the US Remains Unchanged Through. Diabetes Ther. 2017, 8, 863–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okemah, J.; Peng, J.; Quiñones, M. Addressing Clinical Inertia in Type 2 Diabetes Mellitus: A Review. Adv. Ther. 2018, 35, 1735–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Peñalver, J.J.; Martín-Timón, I.; Sevillano-Collantes, C.; Cañizo-Gómez, F.J. del Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 2016, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Bento, C.F.; Pereira, P.; Seiça, R. Diabetes mellitus: New challenges and innovative therapies. EPMA J. 2010, 1, 138–163. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Nonomura, N. Role of androgen receptor in prostate cancer: A review. World J. Men Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Ferro, M.; Lucarelli, G.; Crocetto, F.; Dolce, P.; Verde, A.; La Civita, E.; Zappavigna, S.; de Cobelli, O.; Di Lorenzo, G.; Facchini, B.A.; et al. First-line systemic therapy for metastatic castration-sensitive prostate cancer: An updated systematic review with novel findings. Crit. Rev. Oncol. Hematol. 2021, 157, 103198. [Google Scholar] [CrossRef]
- Buonerba, C.; Ferro, M.; Dolce, P.; Crocetto, F.; Verde, A.; Lucarelli, G.; Scafuri, L.; Facchini, S.; Vaia, A.; Marinelli, A.; et al. Predictors of efficacy of androgen-receptor-axis-targeted therapies in patients with metastatic castration-sensitive prostate cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2020, 151, 102992. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and Cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Giovannucci, E.; Jeon, J.Y. Diabetes and mortality in patients with prostate cancer: A meta-analysis. Springerplus 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faris, J.E.; Smith, M.R. Metabolic sequelae associated with androgen deprivation therapy for prostate cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, A.; Gao, B.; Yeap, S.H.O.; Wong, M.K.Y.; Ali, S.S.; Gurney, H. Metformin in prostate cancer: Two for the price of one. Ann. Oncol. 2011, 22, 2556–2560. [Google Scholar] [CrossRef]
- Karzai, F.H.; Madan, R.A.; Dahut, W.L. Metabolic syndrome in prostate cancer: Impact on risk and outcomes. Future Oncol. 2016, 12, 1947–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannarella, R.; Condorelli, R.A.; Barbagallo, F.; La Vignera, S.; Calogero, A.E. Endocrinology of the Aging Prostate: Current Concepts. Front. Endocrinol. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of diet and nutrition on prostate cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef] [Green Version]
- Capece, M.; Creta, M.; Calogero, A.; La Rocca, R.; Napolitano, L.; Barone, B.; Sica, A.; Fusco, F.; Santangelo, M.; Dodaro, C.A.; et al. Does physical activity regulate prostate carcinogenesis and prostate cancer outcomes? A narrative review. Int. J. Environ. Res. Public Health 2020, 17, 1441. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, F.; Li, D.; Wang, F.; Zhu, L.; Chen, W.; Ge, J.; An, R.; Zhao, Y. Does physical activity reduce the risk of prostate cancer? A systematic review and meta-analysis. Eur. Urol. 2011, 60, 1029–1044. [Google Scholar] [CrossRef]
- Wright, J.L.; Plymate, S.R.; Porter, M.P.; Gore, J.L.; Lin, D.W.; Hu, E.; Zeliadt, S.B. Hyperglycemia and prostate cancer recurrence in men treated for localized prostate cancer. Prostate Cancer Prostatic Dis. 2013, 16, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, M.T.; Selvin, E.; Barber, J.R.; Platz, E.A.; Joshu, C.E. Hyperglycemia, classified with multiple biomarkers simultaneously in men without diabetes, and risk of fatal prostate cancer. Cancer Prev. Res. 2019, 12, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Association, A.D. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43, S98–S110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niswender, K.D. Basal insulin: Physiology, pharmacology, and clinical implications. Postgrad. Med. 2011, 123, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Di Sebastiano, K.M.; Pinthus, J.H.; Duivenvoorden, W.C.M.; Mourtzakis, M. Glucose impairments and insulin resistance in prostate cancer: The role of obesity, nutrition and exercise. Obes. Rev. 2018, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Huang, H. Interplay Among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1210, pp. 319–331. [Google Scholar]
- Zhao, Y.; Tindall, D.J.; Huang, H. Modulation of androgen receptor by FOXA1 and FOXO1 factors in prostate cancer. Int. J. Biol. Sci. 2014, 10, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.L.; Lee, W.; Williams, E.D.; Lubik, A.A.; Stylianou, N.; Shokoohmand, A.; Lehman, M.L.; Hollier, B.G.; Gunter, J.H.; Nelson, C.C. Insulin enhances migration and invasion in prostate cancer cells by up-regulation of FOXC. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.E.; Gleave, M.E.; Zakikhani, M.; Bell, R.H.; Piura, E.; Vickers, E.; Cunningham, M.; Larsson, O.; Fazli, L.; Pollak, M. Insulin receptor expression by human prostate cancers. Prostate 2009, 69, 33–40. [Google Scholar] [CrossRef]
- Ryan, C.J.; Haqq, C.M.; Simko, J.; Nonaka, D.F.; Chan, J.M.; Weinberg, V.; Small, E.J.; Goldfine, I.D. Expression of insulin-like growth factor-1 receptor in local and metastatic prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2007, 25, 134–140. [Google Scholar] [CrossRef]
- Ahearn, T.U.; Peisch, S.; Pettersson, A.; Ebot, E.M.; Zhou, C.K.; Graff, R.E.; Sinnott, J.A.; Fazli, L.; Judson, G.L.; Bismar, T.A.; et al. Expression of IGF/insulin receptor in prostate cancer tissue and progression to lethal disease. Carcinogenesis 2018, 39, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.F.; Glendorf, T.; Hegelund, A.C.; Lundby, A.; Lützen, A.; Slaaby, R.; Stidsen, C.E. Molecular characterisation of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Varewijck, A.J.; Goudzwaard, J.A.; Brugts, M.P.; Lamberts, S.W.J.; Hofland, L.J.; Janssen, J.A.M.J.L. Insulin glargine is more potent in activating the human IGF-I receptor than human insulin and insulin detemir. Growth Horm. IGF Res. 2010, 20, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Karlstad, O.; Linde, J.; Vestergaard, P.; Hjellvik, V.; Bazelier, M.; Schmidt, M.; Andersen, M.; Auvinen, A.; Haukka, J.; Furu, K.; et al. Use of Insulin and Insulin Analogs and Risk of Cancer — Systematic Review and Meta-Analysis of Observational Studies. Curr. Drug Saf. 2013, 8, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Yang, L.; He, Z.; Liu, J. Insulin Glargine and Cancer Risk in Patients with Diabetes: A Meta-Analysis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Saboori, S.; Rad, E.Y.; Birjandi, M.; Mohiti, S.; Falahi, E. Serum insulin level, HOMA-IR and prostate cancer risk: A systematic review and meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 110–115. [Google Scholar] [CrossRef]
- Hammarsten, J.; Högstedt, B. Hyperinsulinaemia: A prospective risk factor for lethal clinical prostate cancer. Eur. J. Cancer 2005, 41, 2887–2895. [Google Scholar] [CrossRef]
- Chen, Y.B.; Chen, Q.; Wang, Z.; Zhou, J. Insulin therapy and risk of prostate cancer: A systematic review and meta-analysis of observational studies. PLoS ONE 2013, 8, e81594. [Google Scholar] [CrossRef]
- Sola, D.; Rossi, L.; Schianca, G.P.C.; Maffioli, P.; Bigliocca, M.; Mella, R.; Corlianò, F.; Paolo Fra, G.; Bartoli, E.; Derosa, G. Sulfonylureas and their use in clinical practice. Arch. Med. Sci. 2015, 11, 840–848. [Google Scholar] [CrossRef]
- Abdul, M.; Hoosein, N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Lett. 2002, 186, 99–105. [Google Scholar] [CrossRef]
- Qi, C.; Li, B.; Yang, Y.; Yang, Y.; Li, J.; Zhou, Q.; Wen, Y.; Zeng, C.; Zheng, L.; Zhang, Q.; et al. Glipizide suppresses prostate cancer progression in the TRAMP model by inhibiting angiogenesis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Haring, A.; Murtola, T.J.; Talala, K.; Taari, K.; Tammela, T.L.J.; Auvinen, A. Antidiabetic drug use and prostate cancer risk in the Finnish Randomized Study of Screening for Prostate Cancer. Scand. J. Urol. 2017, 51, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Shiota, M.; Kobayashi, T.; Kashiwagi, E.; Takeuchi, A.; Inokuchi, J.; Tatsugami, K.; Eto, M. Prognostic significance of diabetes mellitus and dyslipidemia in men receiving androgen-deprivation therapy for metastatic prostate cancer. Prostate Int. 2019, 7, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y.; Nomiyama, T.; Kawanami, T.; Hamaguchi, Y.; Terawaki, Y.; Tanaka, T.; Murase, K.; Motonaga, R.; Tanabe, M.; Yanase, T. Combined Treatment with Exendin-4 and Metformin Attenuates Prostate Cancer Growth. PLoS ONE 2015, 10, e0139709. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell. Mol. Life Sci. 2010, 67, 3407–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Yanase, T.; Morinaga, H.; Okabe, T.; Nomura, M.; Daitoku, H.; Fukamizu, A.; Kato, S.; Takayanagi, R.; Nawata, H. Insulin-like Growth Factor 1/Insulin Signaling Activates Androgen Signaling through Direct Interactions of Foxo1 with Androgen Receptor. J. Biol. Chem. 2007, 282, 7329–7338. [Google Scholar] [CrossRef] [Green Version]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanigur Sultuybek, G.; Soydas, T.; Yenmis, G. NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheirandish, M.; Mahboobi, H.; Yazdanparast, M.; Kamal, W.; Kamal, M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018, 19, 793–797. [Google Scholar] [CrossRef]
- Preston, M.A.; Riis, A.H.; Ehrenstein, V.; Breau, R.H.; Batista, J.L.; Olumi, A.F.; Mucci, L.A.; Adami, H.O.; Sørensen, H.T. Metformin use and prostate cancer risk. Eur. Urol. 2014, 66, 1012–1020. [Google Scholar] [CrossRef]
- Margel, D.; Urbach, D.; Lipscombe, L.L.; Bell, C.M.; Kulkarni, G.; Austin, P.C.; Fleshner, N. Association Between Metformin Use and Risk of Prostate Cancer and Its Grade. J. Natl. Cancer Inst. 2013, 105, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Azoulay, L.; Dell’Aniello, S.; Gagnon, B.; Pollak, M.; Suissa, S. Metformin and the incidence of prostate cancer in patients with type 2 diabetes. Cancer Epidemiol. Biomark. Prev. 2011, 20, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Yin, L.; Jiang, X.; Sun, X.; Wu, J.; Tian, H.; Gao, X.; He, X. Effect of Metformin on Cancer Risk and Treatment Outcome of Prostate Cancer: A Meta-Analysis of Epidemiological Observational Studies. PLoS ONE 2014, 9, e116327. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Yang, Y.; Tang, X.; Skrip, L.; Qiu, J.; Wang, Y.; Zhang, F. Association between metformin therapy and incidence, recurrence and mortality of prostate cancer: Evidence from a meta-analysis. Diabetes Metab. Res. Rev. 2015, 31, 595–602. [Google Scholar] [CrossRef]
- Ghiasi, B.; Sarokhani, D.; Najafi, F.; Motedayen, M.; Dehkordi, A.H. The Relationship Between Prostate Cancer and Metformin Consumption: A Systematic Review and Meta-analysis Study. Curr. Pharm. Des. 2019, 25, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zhou, X.; Liu, N.; Wang, J.; Chen, X.; Xu, X. Metformin use and prostate cancer risk: A meta-analysis of cohort studies. Medicine 2019, 98, e14955. [Google Scholar] [CrossRef]
- He, K.; Hu, H.; Ye, S.; Wang, H.; Cui, R.; Yi, L. The effect of metformin therapy on incidence and prognosis in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, X.; Yan, P.; Tang, J.; Chen, T.; Sun, Y.; Zhou, W.; Bi, Y.; Zhang, Z.J. Effect of metformin on the risk of prostate cancer in patients with type 2 diabetes by considering different confounding factors: A meta-analysis of observational studies. Eur. J. Cancer Prev. 2020, 29, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Eskin, M.; Eurich, D.T.; Majumdar, S.R.; Johnson, J.A. Metformin, Asian ethnicity and risk of prostate cancer in type 2 diabetes: A systematic review and meta-analysis. BMC Cancer 2018, 18, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raval, A.D.; Thakker, D.; Vyas, A.; Salkini, M.; Madhavan, S.; Sambamoorthi, U. Impact of metformin on clinical outcomes among men with prostate cancer: A systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2015, 18, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, L.; Mei, Z.; Xu, C.; Liu, C.; Chu, X.; Hao, B. The impact of metformin use on survival in prostate cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 100449–100458. [Google Scholar] [CrossRef] [Green Version]
- Stopsack, K.H.; Ziehr, D.R.; Rider, J.R.; Giovannucci, E.L. Metformin and prostate cancer mortality: A meta-analysis. Cancer Causes Control 2016, 27, 105–113. [Google Scholar] [CrossRef]
- Hwang, I.C.; Park, S.M.; Shin, D.; Ahn, H.Y.; Rieken, M.; Shariat, S.F. Metformin association with lower prostate cancer recurrence in type 2 diabetes: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2015, 16, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zingales, V.; Distefano, A.; Raffaele, M.; Zanghi, A.; Barbagallo, I.; Vanella, L. Metformin: A Bridge between Diabetes and Prostate Cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Whitburn, J.; Edwards, C.M.; Sooriakumaran, P. Metformin and Prostate Cancer: A New Role for an Old Drug. Curr. Urol. Rep. 2017, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.; Gandhi, J.; Joshi, G.; Smith, N.L.; Khan, S.A. The anticancer potential of metformin on prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ke, R.; Lin, H.; Ying, Y.; Liu, D.; Luo, Z. Metformin, an Old Drug, Brings a New Era to Cancer Therapy. Cancer J. 2015, 21, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, D.; Chamberlain, F.; Garmo, H.; Rudman, S.; Zethelius, B.; Holmberg, L.; Adolfsson, J.; Stattin, P.; Carroll, P.; Van Hemelrijck, M. A systematic review of the literature exploring the interplay between prostate cancer and type two diabetes mellitus. Ecancermedicalscience 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillessen, S.; Gilson, C.; James, N.; Adler, A.; Sydes, M.R.; Clarke, N. Repurposing Metformin as Therapy for Prostate Cancer within the STAMPEDE Trial Platform. Eur. Urol. 2016, 70, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Crawley, D.; Chandra, A.; Loda, M.; Gillett, C.; Cathcart, P.; Challacombe, B.; Cook, G.; Cahill, D.; Santa Olalla, A.; Cahill, F.; et al. Metformin and longevity (METAL): A window of opportunity study investigating the biological effects of metformin in localised prostate cancer. BMC Cancer 2017, 17, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteucci, E.; Giampietro, O. Dipeptidyl Peptidase-4 (CD26): Knowing the Function before Inhibiting the Enzyme. Curr. Med. Chem. 2009, 16, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Haller, R.; Li, S.Y.; Slaton, J.W.; Sinha, A.A.; Wasserman, N.F. Elevation of dipeptidylpeptidase iv activities in the prostate peripheral zone and prostatic secretions of men with prostate cancer: Possible prostate cancer disease marker. J. Urol. 2005, 174, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Wesley, U.V.; McGroarty, M.; Homoyouni, A. Dipeptidyl Peptidase Inhibits Malignant Phenotype of Prostate Cancer Cells by Blocking Basic Fibroblast Growth Factor Signaling Pathway. Cancer Res. 2005, 65, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.-H. Sitagliptin may reduce prostate cancer risk in male patients with type 2 diabetes. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Shah, C.; Hong, Y.R.; Bishnoi, R.; Ali, A.; Skelton, W.P.; Dang, L.H.; Huo, J.; Dang, N.H. Impact of DPP4 Inhibitors in Survival of Patients With Prostate, Pancreas, and Breast Cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Shao, S.; Xu, Q.Q.; Yu, X.; Pan, R.; Chen, Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol. Ther. 2020, 209, 107503. [Google Scholar] [CrossRef]
- Barreira da Silva, R.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, A.; Lawlor, K.; Yi, S.S.; Philip, J.; Ghosh, M.; Yaneva, M.; Villanueva, J.; Saghatelian, A.; Assel, M.; Vickers, A.J.; et al. Inhibition of Circulating Dipeptidyl Peptidase 4 Activity in Patients with Metastatic Prostate Cancer. Mol. Cell. Proteomics 2014, 13, 3082–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-X.; Pedersen, E.A.; Shiozawa, Y.; Havens, A.M.; Jung, Y.; Wang, J.; Pienta, K.J.; Taichman, R.S. CD26/dipeptidyl peptidase IV regulates prostate cancer metastasis by degrading SDF-1/CXCL. Clin. Exp. Metastasis 2008, 25, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.W.; Gao, C.; Bhasin, S.S.; Voznesensky, O.S.; Calagua, C.; Arai, S.; Nelson, P.S.; Montgomery, B.; Mostaghel, E.A.; Corey, E.; et al. Downregulation of Dipeptidyl Peptidase 4 Accelerates Progression to Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 6354–6362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathmann, W.; Kostev, K. Association of dipeptidyl peptidase 4 inhibitors with risk of metastases in patients with type 2 diabetes and breast, prostate or digestive system cancer. J. Diabetes Complicat. 2017, 31, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Vilsbøll, T.; Deacon, C.F. The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 2009, 297, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.J. The Physiology of Glucagon-like Peptide. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Vangoitsenhoven, R.; Mathieu, C.; Van Der Schueren, B. GLP1 and cancer: Friend or foe? Endocr. Relat. Cancer 2012, 19, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Nachnani, J.S.; Bulchandani, D.G.; Nookala, A.; Herndon, B.; Molteni, A.; Pandya, P.; Taylor, R.; Quinn, T.; Weide, L.; Alba, L.M. Biochemical and histological effects of exendin-4 (exenatide) on the rat pancreas. Diabetologia 2010, 53, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, L.B.; Madsen, L.W.; Andersen, S.; Almholt, K.; De Boer, A.S.; Drucker, D.J.; Gotfredsen, C.; Egerod, F.L.; Hegelund, A.C.; Jacobsen, H.; et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010, 151, 1473–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, X.; Chai, S.; Zhao, X.; Ji, L. Risk of Malignant Neoplasia With Glucagon-Like Peptide-1 Receptor Agonist Treatment in Patients With Type 2 Diabetes: A Meta-Analysis. J. Diabetes Res. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yang, S.; Zhou, Z. GLP-1 receptor agonists and risk of cancer in type 2 diabetes: An updated meta-analysis of randomized controlled trials. Endocrine 2019, 66, 157–165. [Google Scholar] [CrossRef]
- Nauck, M.A.; Jensen, T.J.; Rosenkilde, C.; Calanna, S.; Buse, J.B. Neoplasms Reported With Liraglutide or Placebo in People With Type 2 Diabetes: Results from the LEADER Randomized Trial. Diabetes Care 2018, 41, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Nomiyama, T.; Kawanami, T.; Irie, S.; Hamaguchi, Y.; Terawaki, Y.; Murase, K.; Tsutsumi, Y.; Nagaishi, R.; Tanabe, M.; Morinaga, H.; et al. Exendin-4, a GLP-1 Receptor Agonist, Attenuates Prostate Cancer Growth. Diabetes 2014, 63, 3891–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, T.; Nomiyama, T.; Kawanami, T.; Hamaguchi, Y.; Horikawa, T.; Tanaka, T.; Irie, S.; Motonaga, R.; Hamanoue, N.; Tanabe, M.; et al. Activation of overexpressed glucagon-like peptide-1 receptor attenuates prostate cancer growth by inhibiting cell cycle progression. J. Diabetes Investig. 2020, 11, 1137–1149. [Google Scholar] [CrossRef]
- He, W.; Li, J. Exendin-4 enhances radiation response of prostate cancer. Prostate 2018, 78, 1125–1133. [Google Scholar] [CrossRef]
- Wenjing, H.; Shao, Y.; Yu, Y.; Huang, W.; Feng, G.; Li, J. Exendin-4 enhances the sensitivity of prostate cancer to enzalutamide by targeting Akt activation. Prostate 2020, 80, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, S.; Montazeri, H.; Tarighi, P. Synergistic anti-tumor effects of Liraglutide, a glucagon-like peptide-1 receptor agonist, along with Docetaxel on LNCaP prostate cancer cell line. Eur. J. Pharmacol. 2020, 878, 173102. [Google Scholar] [CrossRef] [PubMed]
- Madunić, I.V.; Madunić, J.; Breljak, D.; Karaica, D.; Sabolić, I. Sodium-glucose cotransporters: New targets of cancer therapy? Arch. Ind. Hyg. Toxicol. 2018, 69, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.; Eckstein, N.; Pfeifer, V.; Mayer, P.; Hass, M.D.S. Efficacy, safety and regulatory status of SGLT2 inhibitors: Focus on canagliflozin. Nutr. Diabetes 2014, 4, e143. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.L. Dapagliflozin efficacy and safety: A perspective review. Ther. Adv. Drug Saf. 2014, 5, 242–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Bollu, L.R.; Su, F.; Gao, G.; Xu, L.; Huang, W.-C.; Hung, M.-C.; Weihua, Z. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate 2013, 73, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Villani, L.A.; Smith, B.K.; Marcinko, K.; Ford, R.J.; Broadfield, L.A.; Green, A.E.; Houde, V.P.; Muti, P.; Tsakiridis, T.; Steinberg, G.R. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 2016, 5, 1048–1056. [Google Scholar] [CrossRef]
- Lin, H.-W.; Tseng, C.-H. A Review on the Relationship between SGLT2 Inhibitors and Cancer. Int. J. Endocrinol. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Tang, H.; Dai, Q.; Shi, W.; Zhai, S.; Song, Y.; Han, J. SGLT2 inhibitors and risk of cancer in type 2 diabetes: A systematic review and meta-analysis of randomised controlled trials. Diabetologia 2017, 60, 1862–1872. [Google Scholar] [CrossRef]
- Dicembrini, I.; Nreu, B.; Mannucci, E.; Monami, M. Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2019, 21, 1871–1877. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Elix, C.C.; Salgia, M.M.; Otto-Duessel, M.; Copeland, B.T.; Yoo, C.; Lee, M.; Tew, B.Y.; Ann, D.; Pal, S.K.; Jones, J.O. Peroxisome proliferator-activated receptor gamma controls prostate cancer cell growth through AR-dependent and independent mechanisms. Prostate 2020, 80, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Mori, Y.; Nagano, A.; Naiki-Ito, A.; Kato, H.; Nagayasu, Y.; Kobayashi, M.; Kuno, T.; Takahashi, S. Pioglitazone, a peroxisome proliferator-activated receptor γ agonist, suppresses rat prostate carcinogenesis. Int. J. Mol. Sci. 2016, 17, 2071. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.N.; Lee, J.M.; Oh, H.; Kim, U.; Ryu, B.; Park, J.H. Troglitazone inhibits the migration and invasion of PC-3 human prostate cancer cells by upregulating E-cadherin and glutathione peroxidase. Oncol. Lett. 2018, 16, 5482–5488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Ren, Y.; Chen, A.M.; Guo, F.J.; Xu, F.; Gong, C.; Cheng, P.; Du, Y.; Liao, H. Peroxisome proliferator-activated receptor γ ligands inhibit VEGF-mediated vasculogenic mimicry of prostate cancer through the AKT signaling pathway. Mol. Med. Rep. 2014, 10, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Gong, C.; Chen, A.M.; Guo, F.J.; Xu, F.; Ren, Y.; Liao, H. Peroxisome proliferator-activated receptor γ agonist rosiglitazone inhibits migration and invasion of prostate cancer cells through inhibition of the CXCR4/CXCL12 axis. Mol. Med. Rep. 2014, 10, 695–700. [Google Scholar] [CrossRef]
- Lin, S.J.; Lin, C.Y.; Yang, D.R.; Izumi, K.; Yan, E.; Niu, X.; Chang, H.C.; Miyamoto, H.; Wang, N.; Li, G.; et al. The Differential Effects of Anti-Diabetic Thiazolidinedione on Prostate Cancer Progression Are Linked to the TR4 Nuclear Receptor Expression Status. Neoplasia 2015, 17, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.J.; Yang, D.R.; Li, G.; Chang, C. TR4 nuclear receptor different roles in prostate cancer progression. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Wang, M.; Ding, X.; Xia, L.; Chen, B.; Chen, Y.; Zhang, Z.; Niu, Y.; Li, G.; Chang, C. Testicular orphan nuclear receptor 4 is associated with the radio-sensitivity of prostate cancer. Prostate 2015, 75, 1632–1642. [Google Scholar] [CrossRef]
- Olokpa, E.; Moss, P.E.; Stewart, L.V. Crosstalk between the Androgen Receptor and PPAR Gamma Signaling Pathways in the Prostate. PPAR Res. 2017, 2017, 9456020. [Google Scholar] [CrossRef] [PubMed]
- Moss, P.E.; Lyles, B.E.; Stewart, L.M.V. The PPARγ ligand ciglitazone regulates androgen receptor activation differently in androgen-dependent versus androgen-independent human prostate cancer cells. Exp. Cell Res. 2010, 316, 3478–3488. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, E.; Harding, S.; Lam, H.; Perez, A. Ten-year observational follow-up of PROactive: A randomized cardiovascular outcomes trial evaluating pioglitazone in type 2 diabetes. Diabetes Obes. Metab. 2016, 18, 266–273. [Google Scholar] [CrossRef]
- Lewis, J.D.; Habel, L.A.; Quesenberry, C.P.; Strom, B.L.; Peng, T.; Hedderson, M.M.; Ehrlich, S.F.; Mamtani, R.; Bilker, W.; Vaughn, D.J.; et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA J. Am. Med. Assoc. 2015, 314, 265–277. [Google Scholar] [CrossRef]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after ischemic stroke or Transient Ischemic attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef]
- Kao, L.T.; Xirasagar, S.; Lin, H.C.; Huang, C.Y. Association Between Pioglitazone Use and Prostate Cancer: A Population-Based Case-Control Study in the Han Population. J. Clin. Pharmacol. 2019, 59, 344–349. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knura, M.; Garczorz, W.; Borek, A.; Drzymała, F.; Rachwał, K.; George, K.; Francuz, T. The Influence of Anti-Diabetic Drugs on Prostate Cancer. Cancers 2021, 13, 1827. https://doi.org/10.3390/cancers13081827
Knura M, Garczorz W, Borek A, Drzymała F, Rachwał K, George K, Francuz T. The Influence of Anti-Diabetic Drugs on Prostate Cancer. Cancers. 2021; 13(8):1827. https://doi.org/10.3390/cancers13081827
Chicago/Turabian StyleKnura, Miłosz, Wojciech Garczorz, Adam Borek, Franciszek Drzymała, Krystian Rachwał, Kurian George, and Tomasz Francuz. 2021. "The Influence of Anti-Diabetic Drugs on Prostate Cancer" Cancers 13, no. 8: 1827. https://doi.org/10.3390/cancers13081827