1. Introduction
Prostate cancer (PCa; International Classification of Diseases, ICD: C61) remains a principal cause of cancer-related morbidity and mortality. Globally, 1,414,259 newly diagnosed cases and 375,304 PCa-specific deaths were reported in the year 2020 alone, with a projected 1.72-fold increase in incidence and 1.97-fold increase in mortality worldwide by the year 2040 [
1]. This may be associated with enhanced androgenic signaling, ensuing castration resistance and metastasis, a triad that continues to pose a significant challenge in urology clinics, especially impeding therapeutic success in spite of diagnostic and therapeutic advances [
2,
3,
4]. Current data indicate that at least one in every three PCa cases will acquire a metastatic and/or recurrent phenotype within 48 months of the initial diagnosis [
2,
3]. One-fifth of these patients, with metastatic or recurrent disease, subsequently progress to castration-resistant PCa (CRPC) by the fifth year of clinical follow-up [
2,
3]. Unfortunately, after the development of castration resistance, the approximate median survival is a dismal 14 months [
2,
3].
Cumulative evidence indicates that androgen signaling plays a critical role in the pathogenesis and progression of prostate cancer [
4]. Typically, bio-cellular events that facilitate the viability, survival, and proliferation of cancerous prostate cells are regulated by the androgen receptor (AR) directly or through the modulation of molecular mediators and downstream effectors [
5]. Thus, the mainstay of treatment for metastatic PCa remains androgen-deprivation therapy (ADT), which inhibits AR activity, suppresses AR target genes, and elicits clinical remission that will last a couple of years [
4,
5]. However, because of the non-curative nature of ADT, progression to CRPC often follows, largely secondary to reactivated/restored androgen signaling [
4,
5]. To address this biological phenomenon, second-generation AR inhibitors (ARIs), such as abiraterone and enzalutamide, were developed to further suppress residual AR signaling and are the treatments of choice for patients with CRPC [
6,
7]. However, the clinical conundrum lingers, with a lack of durable complete remission (CR) and eventual treatment failure, despite an initial response and the promise of extended survival [
6,
7].
The early identification of patients at high risk of metastasis and/or recurrence after initial treatment may benefit clinical decision making and aid in the development of an effective treatment strategy that can improve prognosis. Over the past two decades, there has been an increase in biomarker exploration in the field of genitourinary oncology, with piqued interest in the molecular mechanisms underlying the roles of these biomarkers in oncogenicity, metabolic reprogramming, disease progression, and responses to therapy in patients with PCa [
4,
5]. This molecular renaissance, hinged on the discovery and validation of novel diagnostic or prognostic biomarkers, continues to facilitate the elucidation of the pathogenesis and biology of PCa and concomitantly facilitates patient stratification into responders or non-responders to certain therapies, as well as discriminating ‘progressors’ from ‘non-progressors’ [
4,
5,
6,
7]. Against the background of the unabated incidence, high mortality burden, and increased odds of disease progression, as well as a largely unclear underlying bio-mechanism of progression, the present study probed actionable biomarkers that could provide clinically objective measures of PCa biology, improve patient stratification, and inform therapeutic decision making.
Recently, it was reported that the relatively unknown genetic suppressor element 1 (GSE1, KIAA0182), a proline-rich protein with coiled-coil domains, was overexpressed in patients with breast cancer and associated with poor prognosis; targeting GSE1 elicited the upregulation of miR-489-5p, with the repression of breast cancer cell proliferation, migration, and invasion [
8]. In another recent study, it was shown that the expression of GSE1 was upregulated in patients with gastric cancer and, concomitant with enhanced SLC7A5 expression, was implicated in increased tumor growth, metastasis, trastuzumab resistance, and worsened postoperative survival outcomes [
9,
10]. Our evolving understanding of the oncogenic activity of GSE1, coupled with its largely unexplored role in the metastasis, therapy resistance, and susceptibility to disease recurrence in PCa patients, informed the present study.
The last decade has been characterized by the increased evaluation of the biomolecular role of the 40 kDa tumor-associated calcium signal transducer 2 (TACSTD2, also known as trophoblast cell surface antigen 2, TROP2) in tumor initiation and progression. The intron-deficient, epithelial cell adhesion molecule TACSTD2 is a ubiquitously expressed glycoprotein, and its expression is associated with stem cell-defining attributes, including ‘regenerative capacity in various tissues’ [
11,
12,
13].
The epithelium of the adult prostate contains three distinct cell types: basal, luminal, and neuroendocrine [
14]. In this context, there is accruing evidence of the tissue regenerative activity of CD49f + Sca1 + basal cells from the tripartite adult prostate epithelium; interestingly, TACSTD2 is enriched in these basal cells and exhibits stem cell-/progenitor cell-like traits, such as ‘localization to the region of the gland proximal to the urethra and enrichment for sphere-forming and colony-forming cells’ [
14]. However, while the overexpression of TACSTD2 has been described in several cancer types, conflicting reports abound, with functional studies showing not only oncogenic but also tumor suppressor roles [
11]. Interestingly, despite this implication of TACSTD2 in pluripotency and contemporary knowledge that TACSTD2
high basal cells efficiently form spheres in vitro, the role of TACSTD2 in prostate cancerization and therapy response is under-explored or, rather, unclear.
Herein, we present preclinical evidence of the role of GSE1/TACSTD2 in signaling as an indicator of disease course and as a putative biomarker of the therapy responses in patients with PCa. These findings also suggest the clinical feasibility of targeting GSE1 as an efficacious therapeutic strategy to re-sensitize metastatic/recurrent CRPC cells to ADT or as antiandrogen therapy (abiraterone or enzalutamide) while mitigating susceptibility to disease recurrence.
2. Material and Methods
2.1. Prostate Cancer Tissue Samples
Prostate cancer tissue samples (n = 56) were obtained from the Taipei Medical University Shuang Ho Hospital tissue bank, following ethical approval from the Institutional Review Board of the Taipei Medical University (approval number: N202101071). The requirement for patients’ signed informed consent was waived because of the retrospective nature of the study.
2.2. Cell Culture
The normal human primary prostate epithelial HPrEC (ATCC® PCS-440-010™) cell line, LNCaP (ATCC® CRL-1740™; 5-a dihydrotestosterone-responsive, androgen-dependent, and metastatic), PC-3 (ATCC® CRL-1435™; low acid phosphatase and testosterone-5-a reductase, androgen-independent, and metastatic), and the hormone-insensitive DU145 (ATCC® HTB-81™) prostate carcinoma cell line were obtained from the ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were cultured in RPMI 1640 (Thermo Fisher Scientific Inc., Bartlesville, OK, USA), supplemented with 10% fetal bovine serum (FBS, #26140079, Thermo Fisher Scientific Inc., Bartlesville, OK, USA) and 100 U/mL penicillin–streptomycin (Thermo Fisher Scientific Inc., Bartlesville, OK, USA). The cells were subcultured at ≥98% confluence, and the growth media were changed every 72 h.
2.3. Antibodies and Reagents
Monoclonal antibodies against GSE1 (#sc-514946), TACSTD2 (#sc-376746), vimentin (#sc-66002), SLUG/SNAI2 (#sc-166476), BAX (#sc-7480), BCL2 (#sc-7382), VEGF (#sc-7269), OCT3/4 (#sc-5279), MDR1/ABCB1 (#sc-13131), and GAPDH (#sc-32233) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Abiraterone (CB-7598, #S1123, ≥99% (HPLC)) and enzalutamide (MDV3100, #S1250, ≥99% (HPLC)) were purchased from Selleck Chemicals (Houston, TX, USA). Stock solutions of 100 mM in 0.01% dimethyl sulfoxide (DMSO, #276855, Sigma-Aldrich®, Merck KGaA, Darmstadt, Germany) were stored at −20 °C until use.
2.4. Knockdown of GSE1 by shRNA Interference
A GSE1 shRNA plasmid (h) (#sc-93036-SH, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was used to knock down GSE1 in cells, strictly following the manufacturer’s protocol. Briefly, after growing PC-3 or DU145 cells to 70% confluence in 6-well plates, 1 µg/10 µL of re-suspended GSE1shRNA plasmid DNA was diluted in 90 µL of antibiotic-free shRNA Plasmid Transfection Medium (#sc-108062, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for each transfection. After 72 h of incubation, stably transfected cells were selected with 2 μg/mL puromycin (#sc-108071, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), as recommended by the manufacturer. GSE1 knockdown in the cells was verified by Western blot analysis.
2.5. Cell Viability and Proliferation Colorimetric Assay
The sulforhodamine B (SRB) assay was performed to assess cell viability. Wild-type (WT) or GSE1-silenced (shGSE1) PC-3, and DU145 cells were seeded at 3 × 103 cells per well in 96-well plates containing complete growth medium with or without the indicated concentration of abiraterone or enzalutamide and incubated in humidified 5% CO2 at 37 °C for 48 h. Thereafter, the cell viability was evaluated following the manufacturer’s instructions. Briefly, WT or shGSE1 cells were fixed with 10% trichloroacetic acid (TCA: #T6399, Sigma-Aldrich®, Merck KGaA, Darmstadt, Germany), carefully washed with ddH2O, and then stained with 0.4:1 (w/v) SRB–acetic acid solution (#230162, Sigma-Aldrich®, Merck KGaA, Darmstadt, Germany). Unbound SRB dye was carefully washed off the cells with 1% acetic acid, the plates were air-dried, and the bound SRB dye was solubilized in 10 mM Tris base (#3163, Tocris Bioscience, Avon, UK). To analyze cell proliferation, we used the Invitrogen alamarBlue™ High-Sensitivity Cell Viability Reagent (#A50100, Thermo Fisher Scientific Inc., Bartlesville, OK, USA), strictly following the manufacturer’s instructions. Briefly, after seeding PC-3 and DU145 WT (or shGSE1) cells in triplicate, with each assay having three biological replicates, at the indicated time point (Day 2), the cells were incubated with alamarBlue™ at 37 °C for 2 h. The number of dye-stained proliferating cells was measured at an absorbance wavelength of 570 nm in a Molecular Devices SpectraMax M3 Multi-Mode Microplate Reader (Molecular Devices LLC., San Jose, CA, USA).
2.6. Cancer Data Set Retrieval
The public and free-access online cancer data repositories used in this study include The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and Cancer Cell Line Encyclopedia (CCLE). The TCGA dataset used was the prostate adenocarcinoma (PRAD) IlluminaHiSeq RNAseq data (
n = 623), which were downloaded and analyzed using the National Cancer Institute Genomic Data Commons Data Portal (
https://portal.gdc.cancer.gov/, accessed on 9 December 2020) and the Oncomine interface (
https://www.oncomine.org/resource/main.html#v:18, accessed on 5 December 2020). The GSE35988 (
n = 122), GSE6099 (
n = 101), GSE32265 (
n = 55), GSE16560 (
n = 281), GSE21887 (
n = 12), GSE21032 (
n = 281), GSE109708 (
n = 8), and GSE104935 (
n = 10) datasets were all retrieved from the GEO online platform (
https://www.ncbi.nlm.nih.gov/geo/, accessed on 9 December 2020).
2.7. Immunohistochemical (IHC) Staining Assay
Immunohistochemical (IHC) analysis was performed on formalin-fixed, paraffin-embedded (FFPE) sections from our PCa cohort, which consisted of samples from patients with different tumor grades (
n = 56; normal-like: Gleason score (GS) ≤ 5; low: GS = 6; medium: GS = 7; high: GS ≥ 8), following ethical approval by the Taipei Medical University Institutional Review Board (approval number: N202101071) and compliant with recommendations from the Declaration of Helsinki for biomedical research involving human subjects. Samples were probed with antibodies against GSE1, TACSTD2, OCT3/4, and ABCB1/MDR1 at 1:200 dilutions, following the standard IHC protocol. The immunoreactivity, based on the total stained area, stained cell count, average size of the stained area, percentage stained area, and perimeter was quantified using the National Institutes of Health ImageJ software version 1.49 (
https://imagej.nih.gov/ij/).
2.8. Western Blotting Assay
Twenty micrograms of WT, or shGSE1 PC-3, and DU145 cell protein samples were separated using 10% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE). The separated proteins were transferred onto polyvinylidene fluoride (PVDF) membranes using the Bio-Rad Mini-Protein electro-transfer system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PVDF membranes were blocked with 5% non-fat milk in Tris-buffered saline with Tween 20 (TBST) for 1 h and then incubated with primary monoclonal antibodies against GSE1 (1:1000, Santa Cruz Biotechnology), TACSTD2 (1:1000, Santa Cruz Biotechnology), vimentin (1:1000, Santa Cruz Biotechnology), SLUG/SNAI2 (1:1000, Santa Cruz Biotechnology), BAX (1:1000, Santa Cruz Biotechnology), BCL2 (1:1000, Santa Cruz Biotechnology), VEGF (1:1000, Santa Cruz Biotechnology), OCT3/4 (1:1000, Santa Cruz Biotechnology), MDR1/ABCB1 (1:1000, Santa Cruz Biotechnology), and GAPDH (1:1000, Santa Cruz Biotechnology) overnight at 4 °C. Thereafter, the PVDF membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature and washed thrice with cold 1X phosphate-buffered saline (PBS, #11666789001, Sigma-Aldrich
®, Merck KGaA, Darmstadt, Germany); then, the protein bands were detected using an enhanced chemiluminescence detection system (Thermo Fisher Scientific Inc., Waltham, MA, USA). Densitometry was performed with the National Institutes of Health ImageJ software version 1.49 (
https://imagej.nih.gov/ij/).
2.9. Scratch Wound-Healing Assay
A scratch wound-healing assay was performed to assess cell migration. Briefly, WT, or shGSE1 PC-3, and DU145 cells were seeded and cultivated in 6-well plates (Corning, Corning, NY, USA) containing complete growth medium with 10% FBS. The medium was changed to low-serum (1% FBS) growth medium when the cells reached >98% confluence. The median axes of the single-layered, adherent cells were scratched using sterile yellow pipette tips. The cell migration, based on the closure of the scratch wounds, was monitored over time, and images were captured at 0 and 24 h post-denudation under a light microscope using a 10X objective lens. Thereafter, the images were analyzed using the National Institutes of Health ImageJ software version 1.49 (
https://imagej.nih.gov/ij/).
2.10. Tumorsphere Formation Assay
WT, or shGSE1 PC-3, and DU145 cells were seeded at 5 × 104 cells per well in ultra-low-attachment 6-well plates (Corning, Corning, NY, USA) containing RPMI 1640, supplemented with 20 ng/mL basic fibroblast growth factor (bFGF; #13256029, Invitrogen), GibcoTM B-27TM supplement (#17504044, Invitrogen, Carlsbad, CA, USA), and 20 ng/mL epidermal growth factor (EGF; #PHG0311, Invitrogen). The PCa cell lines were cultured at 37 °C in a humidified 5% CO2 incubator for 5–7 days. The formed tumorspheres with sizes ≥ 100 µm were counted under an inverted phase-contrast microscope.
2.11. Tumor Xenograft In Vivo Studies
For in vivo tumor xenograft studies, 1 × 106 PC-3_WT, or PC-3_shGSE1, cells in 100 μL of complete growth medium were subcutaneously injected into the right flanks of 7–8-week-old male BALB/c-nu mice (28.3 ± 5.2 g; n = 5 per group) (BioLASCO, Taipei City, Taiwan). The mice were randomly divided into the control (PC-3_WT) and test (PC-3_shGSE1, enzalutamide, and PC-3_shGSE1+enzalutamide) groups. For the treatment group, 10 mg/kg/day of enzalutamide, administered intraperitoneally (ip) every 72 h for 4 weeks, was initiated as soon as the tumors became palpable (tumor volume ~100 mm3). For the control (positive: PC-3_WT, or negative: PC-3_shGSE1) group, 100 μL/day of vehicle 0.01% DMSO was administered ip every 72 h for 4 weeks. Tumor growth was monitored throughout the experiment by taking caliper measurements of tumors twice weekly, and the tumor volume was estimated using the following formula: ½ [length (mm)] × [width (mm)]2. The mice were humanely sacrificed at the end of the study on day 30. The tumors were excised and carefully analyzed, and tumor samples were used for subsequent assays. The animal studies complied with the approved protocol of the Lab Animal Committee/Institutional Animal Care and Use Committee (Approval no.: LAC-2020-0553) of Taipei Medical University.
2.12. Statistical Analysis
All the data are expressed as the mean ± standard deviation (SD) for assays performed at least 3 times independently. Two-sided Student’s t-tests were used for comparison between 2 groups, while one-way ANOVA with Tukey’s post-hoc tests were used for comparing ≥3 groups. Kaplan–Meier survival analyses aided in the comparison of survival rates between the control and test groups. The Pearson chi-square (X2) test was used for correlation analysis and the determination of association. All the statistical analyses were performed using GraphPad Prism version 8.0.0 for Windows (GraphPad Software, La Jolla, CA, USA). The p-values < 0.05 were considered statistically significant.
4. Discussion
Despite significant advances in diagnostic and therapeutic strategies in the last three decades, metastatic castration-resistant prostate cancer (mCRPC) remains a therapeutic enigma. In the last decade alone, the United States Food and Drugs Administration (US FDA) approved six new anticancer drugs for managing CRPC, with a seventh one ascribed a ‘breakthrough designation for accelerated development based on biomarker status’ [
22], highlighting the clinical relevance and therapeutic indispensability of actionable biomarkers in the management of ‘difficult-to-treat’ and ‘quick-to-relapse’ malignancies, such as mCRPC. Premised on this understanding, we aimed to identify such objective indicators of cancerization, disease course, and therapy response in patients with advanced PCa, including mCRPC.
In the present study, we demonstrated (for the first time, to the best of our knowledge) that (i) an increased GSE1/TACSTD2 expression ratio marks patients with PCa and that (ii) the interaction between GSE1 and TACSTD2 drives metastatic disease, castration resistance, and disease progression in patients with PCa. We also provide preclinical evidence that (iii) inversely correlated GSE1 and TACSTD2 expression patterns determine the survival of patients with PCa and that (iv) the GSE1 and TACSTD2 signal interplay affects the clinical and immune statuses and (v) determines the therapy responses and clinical outcomes in patients with PCa. Of therapeutic relevance, we also demonstrate that (vi) the GSE1 and TACSTD2 interaction, or expression profiles, reflect abiraterone/enzalutamide drug resistance, androgen sensitivity, and castration resistance in patients with prostate cancer and posit that (vi) targeting GSE1 signaling suppresses metastatic and cancer stemness phenotypes and enhances sensitivity to abiraterone or enzalutamide in metastatic castration-resistant PCa in vitro and in vivo.
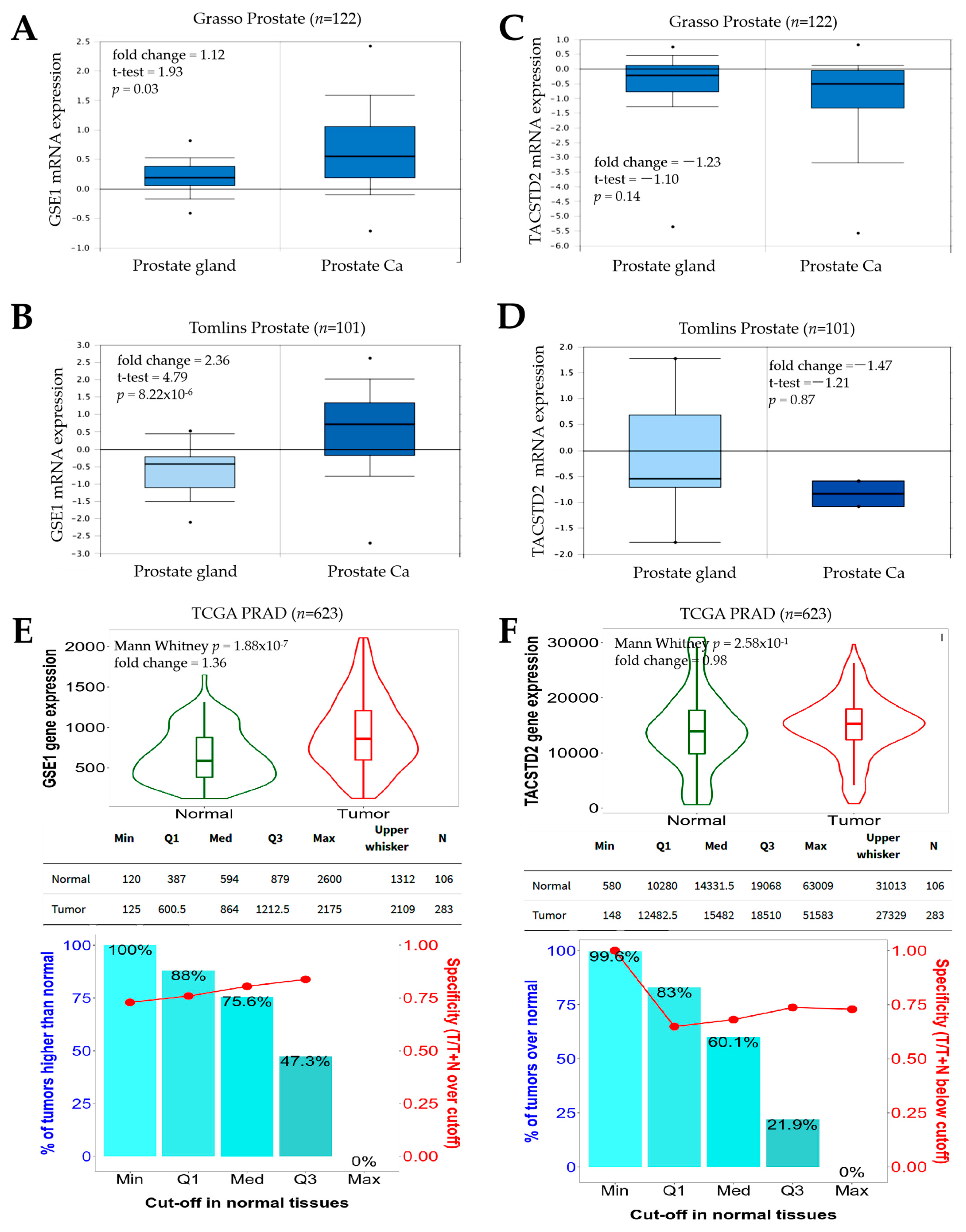
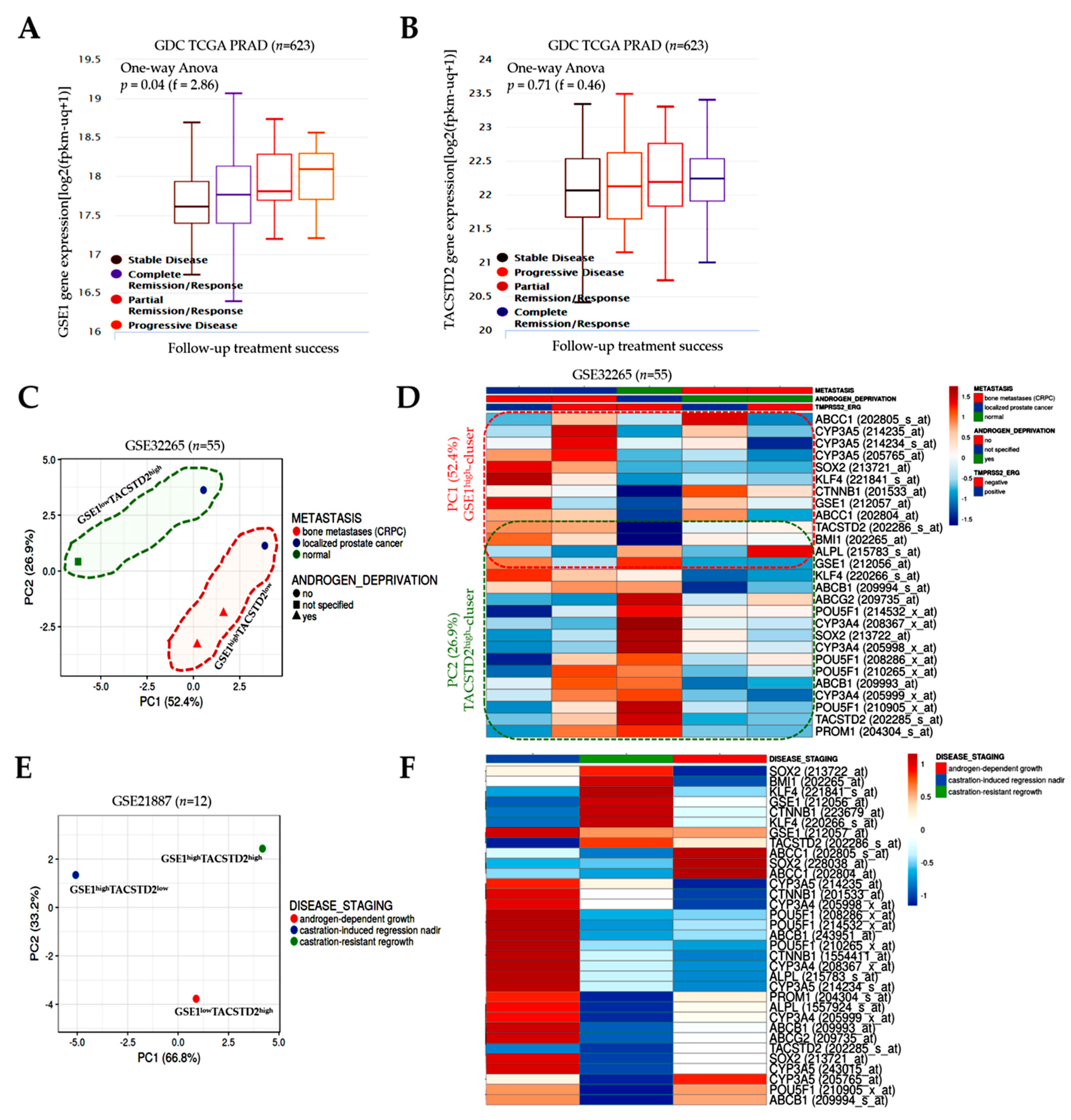
The observation that GSE1 is upregulated, while TACSTD2 is downregulated, in PCa, with an increased GSE1/TACSTD2 expression ratio defining patients with PCa (
Figure 1), is not logically decoupled from the current dysregulated oncogene–tumor suppressor homeostasis paradigm associated with tumor initiation and progression. Our finding is, in part, corroborated by a recent report that the oncoprotein GSE1 is aberrantly expressed in breast cancer and implicated in the proliferation, migration, and invasion of breast cancer cells [
8]. Similar to TACSTD2, the same study also suggested that hsa-miR-489-5p, an onco-miR, is a direct target of GSE1 and exhibits suppressed expression in breast cancer cells [
8].
While this study attributes a tumor suppressor role to TACSTD2 in PCa, we do acknowledge that this finding contradicts the findings of Hsu EC et al., who conversely stated that TACSTD2 is overexpressed in CRPC, drives cancer growth, and induces the neuroendocrine phenotype [
23]. We cannot fully explain this contradiction; however, we cautiously attribute this to the tumor heterogeneity, mutational status, and/or therapeutic context. We find it intriguing that it was posited that the oncogenic activity of TACTSD2 was mediated by the upregulation of PARP1 [
23], considering that the overactivation of the full-length PARP1 induces rapid cellular energy depletion, eliciting a shift in the cell death continuum from apoptosis to necrosis or necroptosis in the presence of enhanced DNA damage [
24]. The ensuing conundrum is that, even in the presence of mild DNA damage, cleaved PARP1 quells necrosis/necroptosis, promotes apoptosis, and prevents cell survival [
24]. Thus, in the context of [
23,
24], the induction or overexpression of TACSTD2 would elicit cell death, consistent with the position of the current study. Aligned with the therapeutic context hypothesis, although PARP1 participates in the DNA repair process, excessive chemotherapeutic- and/or radiation-induced DNA damage prompts PARP1 (and, by inference, TACSTD2) overactivation, adenosine triphosphate (ATP) depletion, and cell death, secondary to bio-energetic collapse [
25,
26]. It is also probable that the tumor suppressor role of TACSTD2, documented in our study, is associated with the resulting suppression of the synthesis of intranucleolar ribosomal RNA and the nuclear translocalization of TACSTD2 in the presence of GSE1 downregulation [
27].
Our present findings provide some rationale for looking outside the contemporary classification of TACSTD2 as an oncogene by highlighting several tumor suppressor traits that make TACSTD2 a putative, actionable biomarker that should be carefully considered when developing any meaningful anti-mCRPC therapeutic strategy. In agreement with Shen et al.’s propositions on ‘genes with both oncogenic and tumor suppressor functions’ [
28], it is probable that non-silent, function-altering mutations, such as frameshift or point mutations in TACSTD2, are the principal drivers of the prevalent tumor-limiting and therapy-sensitizing TACSTD2 signaling in CRPC, documented herein. Interestingly, similar to that of TP53, the mode of TACSTD2 activation is quite unique, compared to that of most other tumor suppressors; 82.5% of TACSTD2 mutations are truncating/nonsense mutations, as a C → T at nucleotide 352 replaces the glutamine at codon 118 with a Q118X stop codon [
29]. This facilitates the synthesis of a stable mutant TACSTD2 protein, which accumulates in the plasma membrane and nucleus, subsequent to the inhibition of GSE1 signaling in aggressive mCRPC cells exposed to antiandrogen therapy and/or ADT. This high frequency of the premature termination/truncation of translation is strongly analogous to “four well-established tumor suppressors (PTEN, TP53, FBXW7, and CDKN2A), and PPP2R1A, a central component of the protein phosphatase 2A (PP2A) complex that also functions as a tumor suppressor” [
30], regardless of the difference in the mutation spectrum. Moreover, while several studies have reported the activation of protein kinase B (Akt) by TACSTD2, mostly in cancer cell lines, several others have also described the downregulation of Akt and MAPK/ERK pathways by TACSTD2 (reviewed in [
11]), thus indicating that the prevalent effect of TACSTD2 may be context-dependent. This rationalization suggests that tumor heterogeneity, the therapeutic context, and the mutational status are therapeutically relevant, as they transcend the initial biological function attributed to TACSTD2 and can inform the discovery or development of new, highly efficacious anti-mCRPC therapeutic strategies.
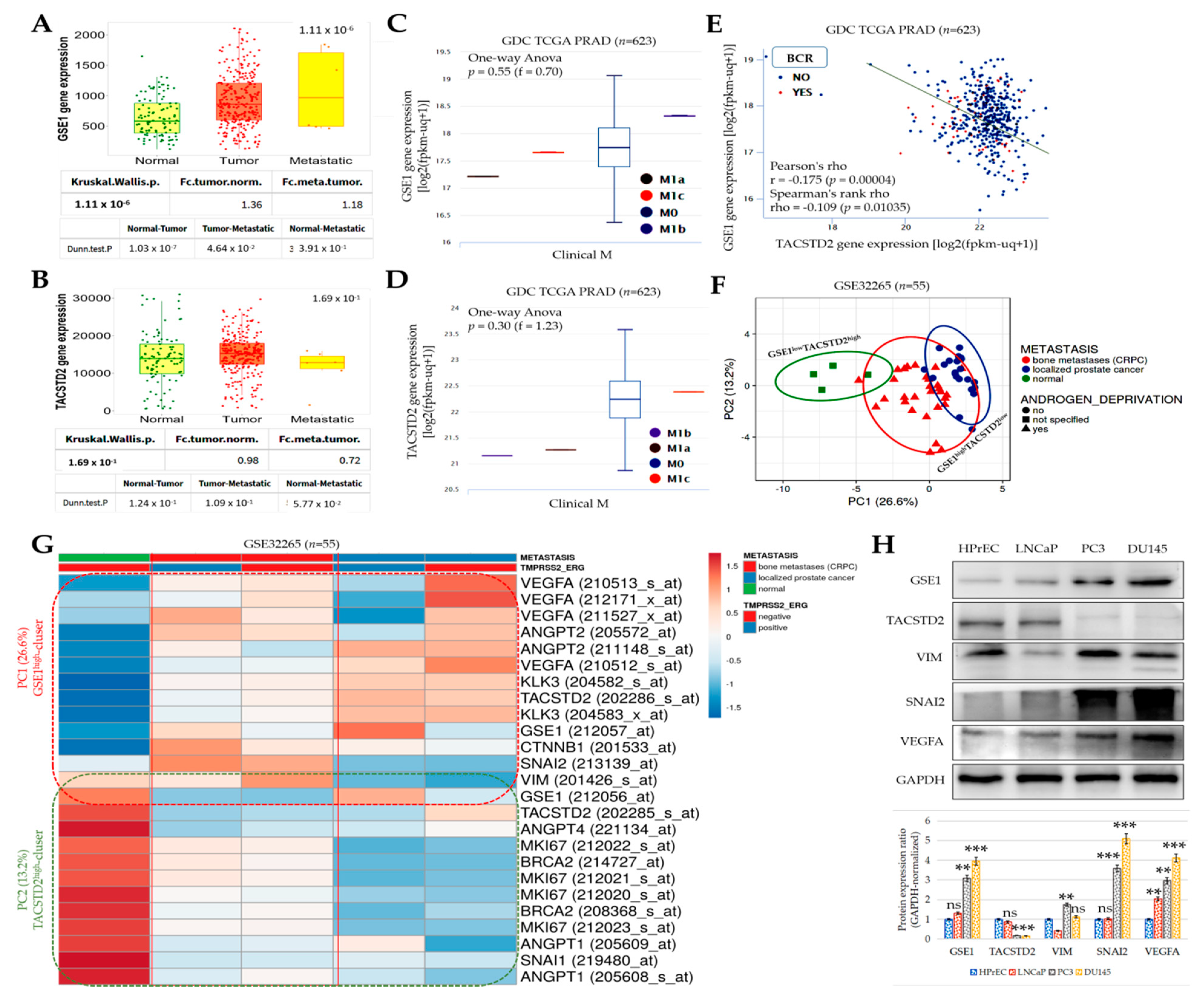
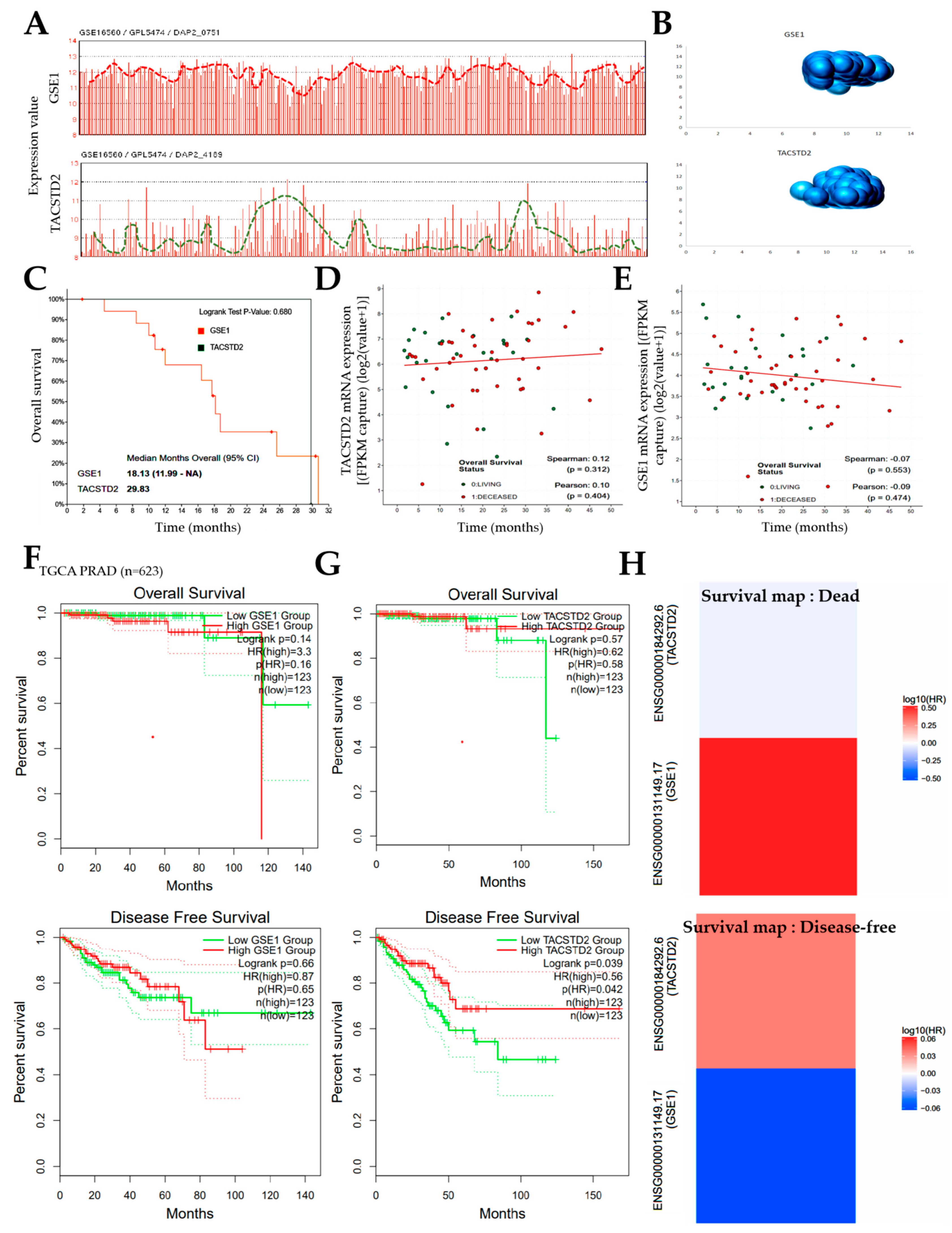
Furthermore, concordant with our finding that the inversely correlated expression of GSE1 and TACSTD2 drives metastatic disease, castration resistance, and disease progression, as well as aids in determining the survival of patients with PCa (
Figure 2 and
Figure 3), some recent studies have indicated that high GSE1 expression was strongly associated with an advanced clinical stage, a high histological grade, a depth of invasion > 5 mm, lymph node metastasis, and decreased sensitivity to trastuzumab, as well as positively correlated with worse survival rates in patients with gastric cancer [
9,
10].
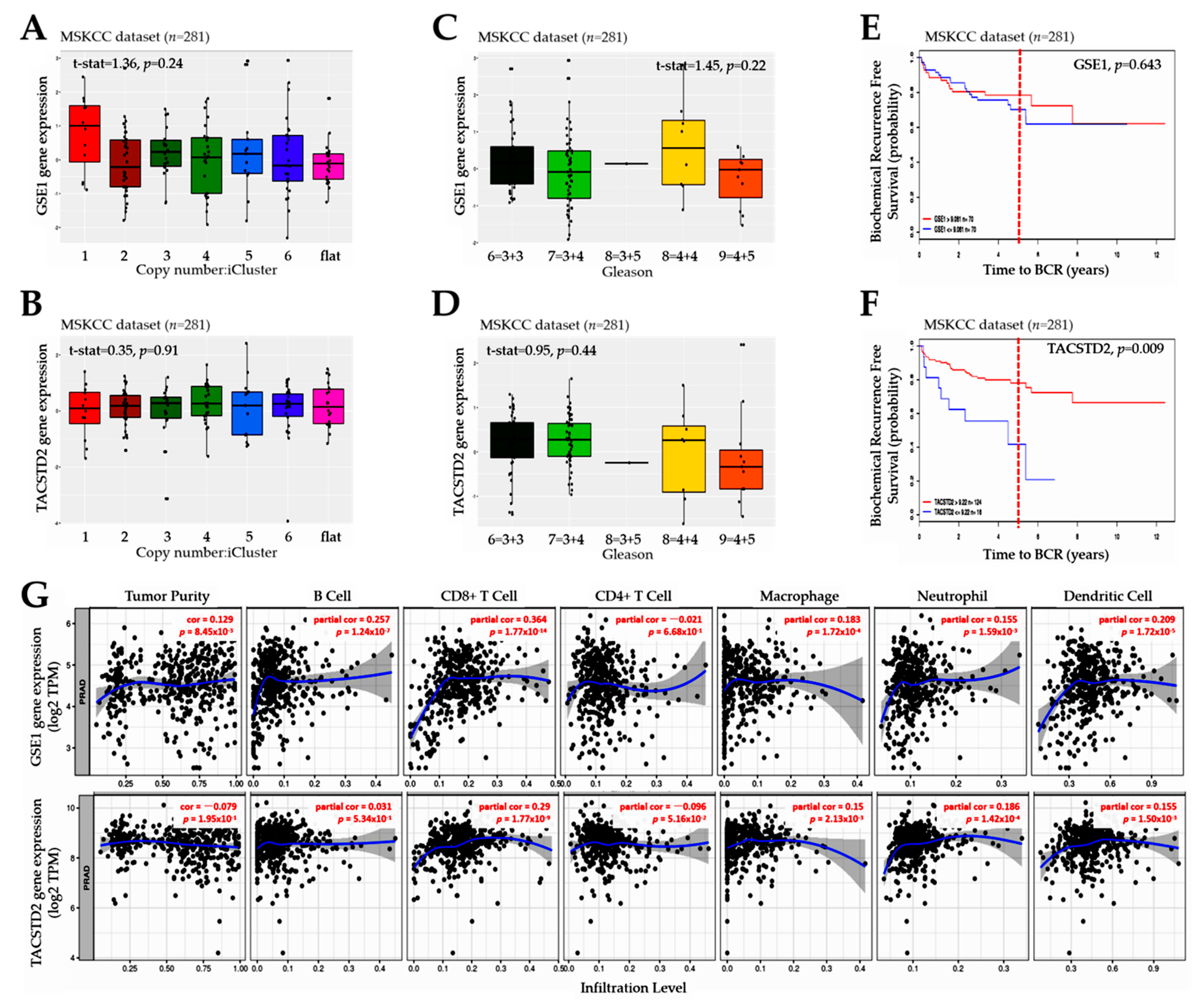
Consistent with contemporary knowledge that molecular interactions induced or facilitated by proline-rich motifs characterize many aspects of the immune response, and that these proline-rich factors mediate cell–cell communication, signal transduction, and antigen recognition [
31], we also demonstrated that the GSE1 and TACSTD2 signal interplay affects the clinical and immune status and is indicative of the therapy responses and clinical outcomes in patients with PCa (
Figure 4 and
Figure 5). It is clinically relevant that
GSE1 expression was higher in iClusters 1, 3, and 5 than in iClusters 2 and 6, while conversely,
TACSTD2 expression was lower in iClusters 1 and 5 than in iClusters 2, 3, 4, and 6. Corroborating this finding, a recent report indicates that, in terms of the time to the BCR endpoint, primary tumors in ‘minimally altered cluster 2 had an extremely favorable prognosis’, in contrast to the ‘extremely unfavorable prognosis for the highly altered cluster 5 tumors’ [
15]. In addition, our finding is consistent with outcome reports from the Cambridge cohort, wherein iCluster 2 and iCluster 1 clearly distinguished patient groups with better and worse prognoses, respectively, based on BCR survival data collected over 60 months [
16]. In fact, the same study reported that ‘iClusters1 and 3 identified men with the highest risk of relapse more effectively than either elevated Gleason score (≥4 + 3), high PSA, extracapsular extension (ECE) or positive surgical margin (PSM), especially as about 6-in-10 of the iCluster1, and 8-in-10 of the iCluster3 patients progressed to recurrent disease’ [
16]. It is, thus, conceivable that an elevated GSE1/TACSTD2 ratio better stratifies patients into good and poor outcome groups than PSA, ECE, and PSM. This study is currently ongoing.
Cancer is widely considered to be a systemic disease that induces a myriad of functional and constitutive changes to the host immune system as a whole. Based on the findings of the present study, we posit that GSE1–TACSTD2 signaling modulates existing host anticancer immunity through the reprogramming of the stromal and intratumoral infiltrating immune cell pool, as we demonstrated, to elicit an immune-excluded/suppressive TME, and subsequent treatment failure (for the GSE1
highTACSTD2
low genotype), or immune-activated/reinvigorated hot tumors, resulting in durable remission (for the GSE1
lowTACSTD2
high genotype) [
32].
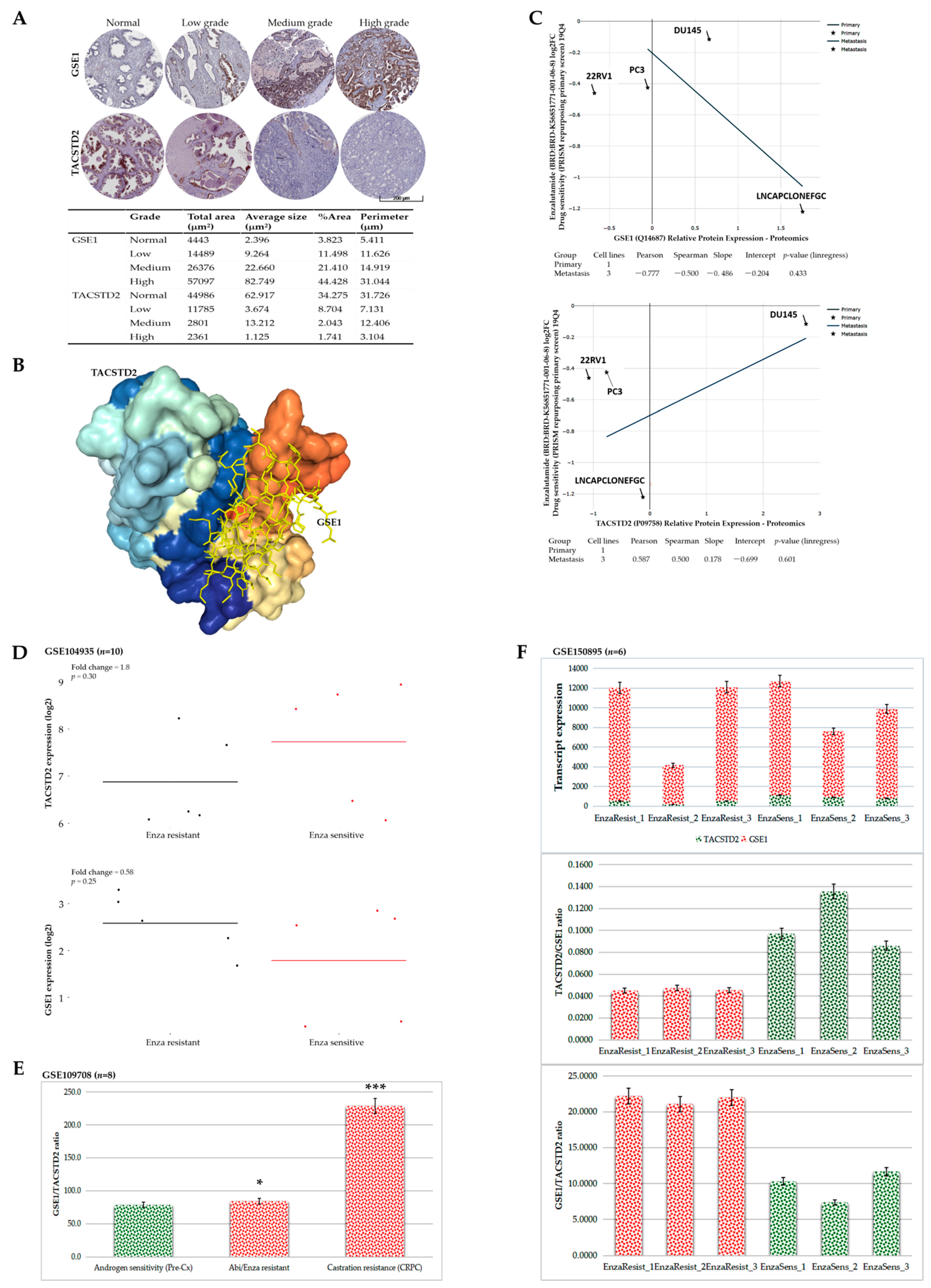
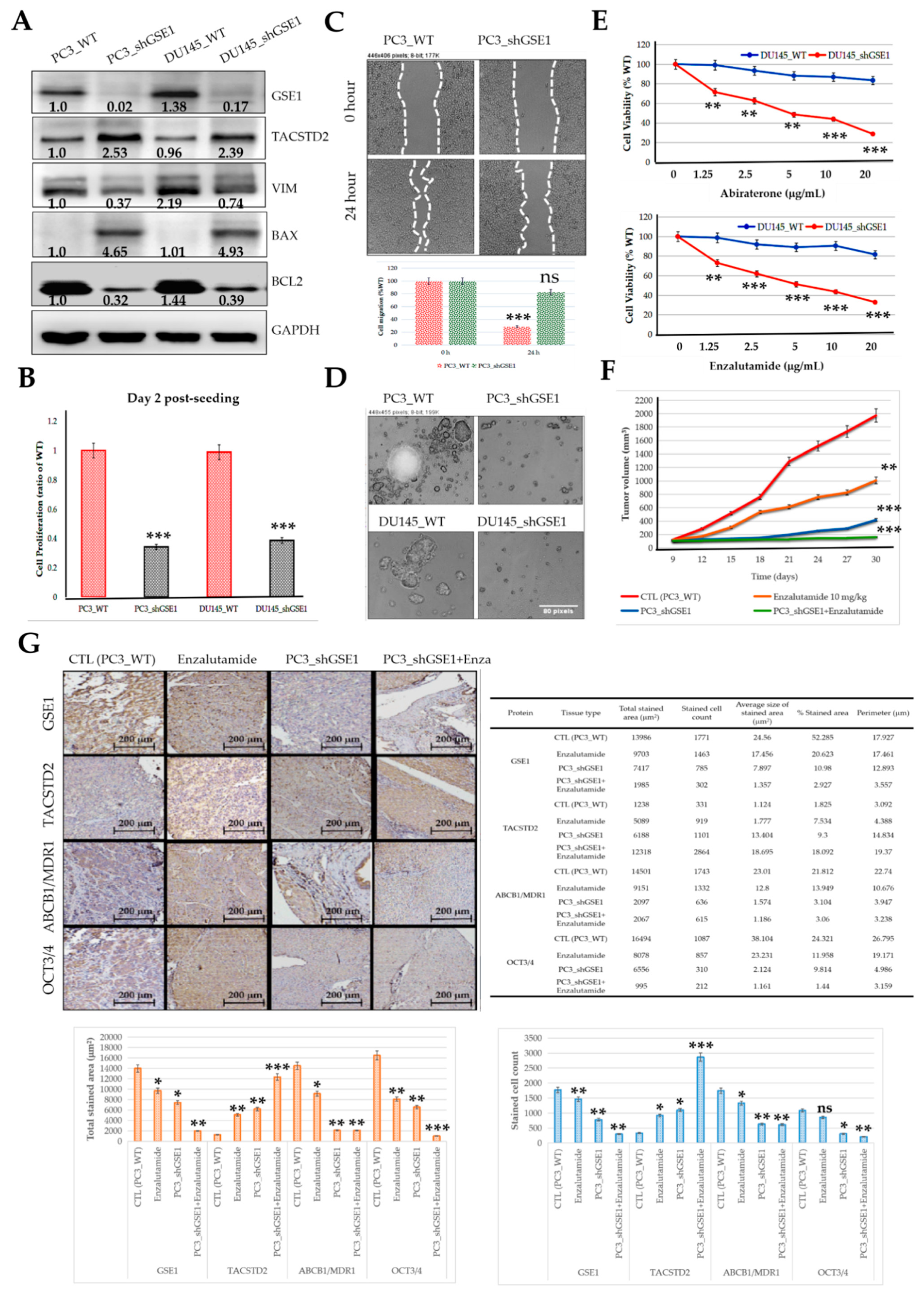
Consistent with their demonstrated role in immunoediting (for the first time, to the best of our knowledge), we also provided some preclinical evidence that the GSE1/TACSTD2 ratio reflects abiraterone/enzalutamide and castration resistance in patients with PCa and that targeting GSE1 signaling suppresses metastatic and cancer stemness phenotypes and enhances sensitivity to abiraterone or enzalutamide in metastatic castration-resistant PCa in vitro and in vivo (
Figure 6 and
Figure 7). This is of translational relevance, especially considering that, despite the touted benefits of enzalutamide and abiraterone, only a subset of patients with CRPC respond to these treatments, with a rather disappointing increase in median PFS of < 6 months, relative to that with the standard of care and an almost universal acquisition of abiraterone or enzalutamide resistance [
33]. In line with the demonstrated
GSE1lowTACSTD2high expression in enzalutamide-sensitive cases,
GSE1highTACSTD2low expression in enzalutamide-resistant cases, and high GSE1/TACSTD2 ratio in resistant cases, we posit that, while the known mechanisms underlying abiraterone or enzalutamide resistance remain inconclusive and continue to evolve, it is probable that by binding to and suppressing the catalytic activity of TACSTD2, GSE1 upregulates intratumoral and/or systemic androgen biosynthesis and deregulates pathways that crosstalk with androgen receptor (AR) signaling, with a consequent amplification of cancer stemness and AR signaling, while concomitantly facilitating an immunosuppressive TME.
As is characteristic of many studies of this nature, the present preclinical study was limited by the need for the inclusion of more clinicopathological features of PCa, such as the Gleason grade, AR score, volume of metastatic disease, microsatellite instability (MSI) score, tumor mutation burden (TMB), and RAF1/BRAF status. As such, the data presented herein should be interpreted ‘as is’ and with cautious optimism.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






