
The Role of MicroRNAs in Lung Cancer Metabolism


Abstract
:Simple Summary
Abstract
1. Introduction
2. MiRNAs Involved in Lung Cancer Metabolism
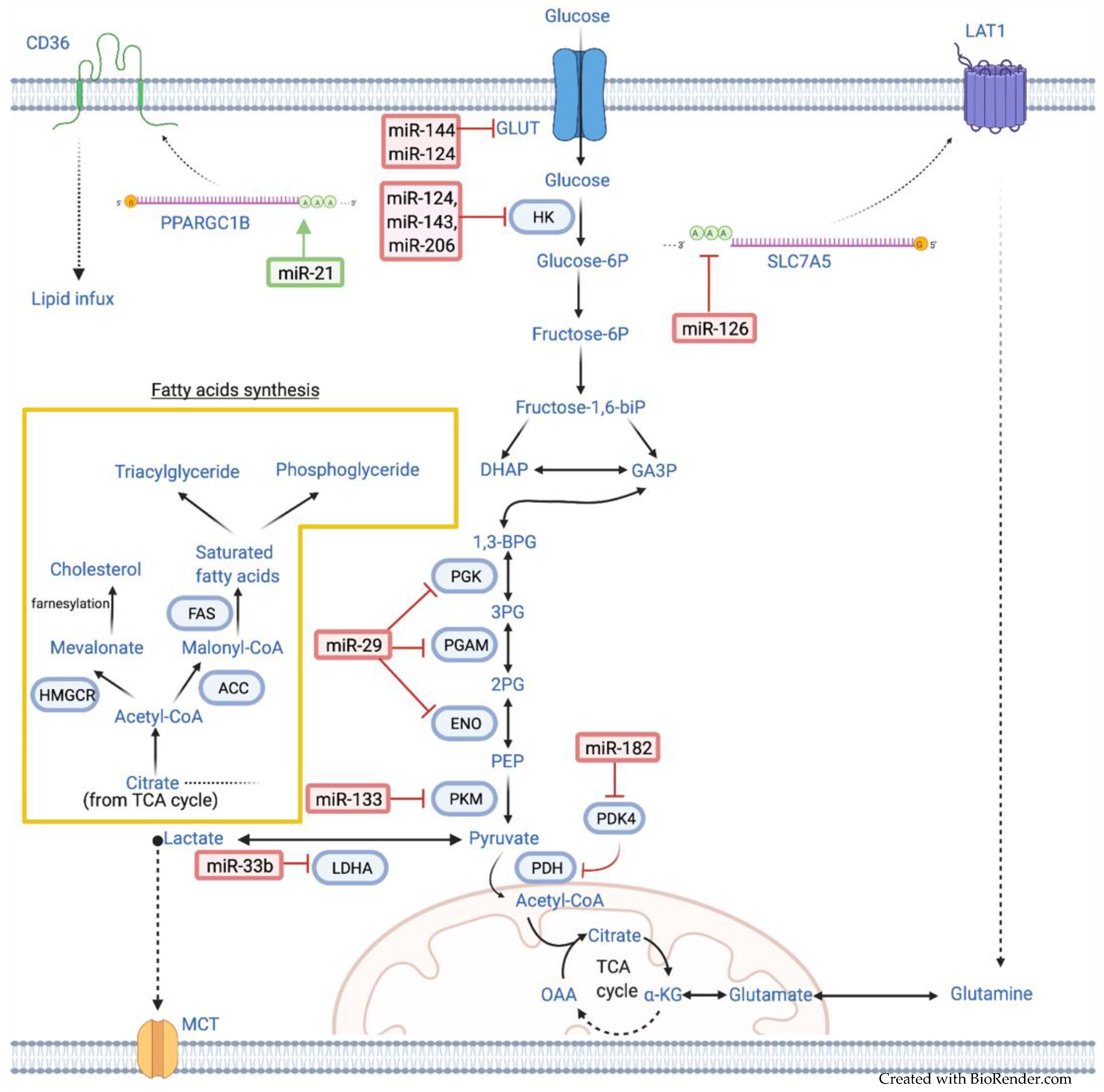
2.1. MiRNAs Associated with Glucose Metabolism
2.1.1. MiR-144
2.1.2. MiR-124
2.1.3. MiR-143, MiR-206, MiR-1
2.1.4. MiR-133
2.1.5. MiR-29
2.2. MiRNAs Associated with Lipid Metabolism
2.2.1. MiR-33b
2.2.2. MiR-21
2.2.3. MiR-182
2.3. MiRNAs Associated with Amino Acid Metabolism
MiR-126
2.4. MiRNAs Involved in the Metabolic Signaling Pathway
2.4.1. PI3k/Akt/PTEN Pathway
2.4.2. c-Myc Pathway
2.4.3. P53 Pathway
3. Relationship between MiRNAs and Hypoxia in Lung Cancer
3.1. MiR-210
3.2. MiR-31-5p
3.3. MiR-199a-5p, MiR-34
3.4. MiR-21, MiR-200c, MiR-519c
4. Future Perspective of MiRNA Targeted Lung Cancer Therapy and Clinical Implication of Existing Chemotherapies on MiRNA Expression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The epidemiology of lung cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Onaitis, M.W.; Hanna, J.M. Cell of origin of lung cancer. J. Carcinog. 2013, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Fang, S.; Gu, W. The Molecular Mechanism of Metabolic Remodeling in Lung Cancer. J. Cancer 2020, 11, 1403–1411. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.; Serpa, J. Metabolic Remodelling: An Accomplice for New Therapeutic Strategies to Fight Lung Cancer. Antioxidants 2019, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, H.; Seto, M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 2005, 19, 2013–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Serra, P.; Esteller, M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene 2011, 31, 1609–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, B.; Stresemann, C.; Brueckner, B.; Lyko, F. Methylation of Human MicroRNA Genes in Normal and Neoplastic Cells. Cell Cycle 2007, 6, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Guo, Y.-N.; Shi, K.; Huang, H.-M.; Huang, S.-P.; Xu, W.-Q.; Li, Z.-Y.; Wei, K.-L.; Gan, T.-Q.; Chen, G. Down-regulation of microRNA-144-3p and its clinical value in non-small cell lung cancer: A comprehensive analysis based on microarray, miRNA-sequencing, and quantitative real-time PCR data. Respir. Res. 2019, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lu, C.; Chu, W.; Zhang, B.; Zhen, Q.; Wang, R.; Zhang, Y.; Li, Z.; Lv, B.; Li, H.; et al. MicroRNA-124 suppresses proliferation and glycolysis in non–small cell lung cancer cells by targeting AKT–GLUT1/HKII. Tumor Biol. 2017, 39, 1010428317706215. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Xiao, T.; Fang, Z.; Sun, Y.; Li, F.; Gao, Y.; Feng, Y.; Li, L.; Wang, Y.; Liu, X.; et al. MicroRNA-143 (miR-143) Regulates Cancer Glycolysis via Targeting Hexokinase 2 Gene. J. Biol. Chem. 2012, 287, 23227–23235. [Google Scholar] [CrossRef] [Green Version]
- Jia, K.-G.; Feng, G.; Tong, Y.-S.; Tao, G.-Z.; Xu, L. miR-206 regulates non-small-cell lung cancer cell aerobic glycolysis by targeting hexokinase 2. J. Biochem. 2019, 167, 365–370. [Google Scholar] [CrossRef]
- Liu, G.; Li, Y.; Gao, X. Overexpression of microRNA-133b sensitizes non-small cell lung cancer cells to irradiation through the inhibition of glycolysis. Oncol. Lett. 2016, 11, 2903–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniyappa, M.; Dowling, P.; Henry, M.; Meleady, P.; Doolan, P.; Gammell, P.; Clynes, M.; Barron, N. MiRNA-29a regulates the expression of numerous proteins and reduces the invasiveness and proliferation of human carcinoma cell lines. Eur. J. Cancer 2009, 45, 3104–3118. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Zhao, L.; Lin, T.; Wang, W. Downregulation of miR-33b promotes non-small cell lung cancer cell growth through reprogramming glucose metabolism miR-33b regulates non-small cell lung cancer cell growth. J. Cell. Biochem. 2019, 120, 6651–6660. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Liu, Y.; Wang, Y.; Meng, M.; Wang, K.; Zang, X.; Zhao, S.; Sun, X.; Cui, L.; Pan, L.; et al. MiR-21 and MiR-155 promote non-small cell lung cancer progression by downregulating SOCS1, SOCS6, and PTEN. Oncotarget 2016, 7, 84508–84519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.; Cheng, X.; Li, Y.; Han, Y.; Song, X.; Yu, D.; Cao, X.; Liu, Z. MiR-21 improves invasion and migration of drug-resistant lung adenocarcinoma cancer cell and transformation of EMT through targetingHBP1. Cancer Med. 2018, 7, 2485–2503. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, M.; Hu, J.; Lei, R.; Xiong, H.; Ji, H.; Yin, H.; Wei, Q.; Hu, G. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene 2016, 36, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Margitai, Z.; Czimmerer, Z.; Várkonyi, I.; Dezso, B.; Lányi, A.; Bacsó, Z.; Scholtz, B. miR-126 inhibits proliferation of small cell lung cancer cells by targeting SLC7A5. FEBS Lett. 2011, 585, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, M.; Liu, J.; Sun, Z.; Ni, J.; Liu, H. miRNA-125b regulates apoptosis of human non-small cell lung cancer via the PI3K/Akt/GSK3β signaling pathway. Oncol. Rep. 2017, 38, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Huang, J.; Zhang, K.; Pan, B.; Chen, J.; De, W.; Wang, R.; Chen, L. MicroRNA-451 induces epithelial–mesenchymal transition in docetaxel-resistant lung adenocarcinoma cells by targeting proto-oncogene c-Myc. Eur. J. Cancer 2014, 50, 3050–3067. [Google Scholar] [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional Activation of miR-34a Contributes to p53-Mediated Apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2009, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Gomez-Arroyo, J.; Alhussaini, A.; Kraskauskas, D.; Cool, C.D.; Voelkel, N.F. MicroRNA-199a-5p Is Associated With Hypoxia-Inducible Factor-1α Expression in Lungs From Patients With COPD. Chest 2012, 142, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puissegur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.A.; Del Grosso, S.J.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2010, 18, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Edmonds, M.D.; Boyd, K.L.; Moyo, T.; Mitra, R.; Duszynski, R.; Arrate, M.P.; Chen, X.; Zhao, Z.; Blackwell, T.S.; Andl, T.; et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J. Clin. Investig. 2015, 126, 349–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.A.; Arora, S.; Prakasam, G.; Calin, G.A.; Syed, M.A. MicroRNA in lung cancer: Role, mechanisms, pathways and therapeutic relevance. Mol. Asp. Med. 2019, 70, 3–20. [Google Scholar] [CrossRef]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhove, K.; Derveaux, E.; Graulus, G.-J.; Mesotten, L.; Thomeer, M.; Noben, J.-P.; Guedens, W.; Adriaensens, P. Glutamine Addiction and Therapeutic Strategies in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 252. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-Q.; Hou, B.-H.; LaLonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.-Q.; Guo, W.-J.; Kim, J.-G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nat. Cell Biol. 2010, 468, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Ancey, P.; Contat, C.; Meylan, E. Glucose transporters in cancer—From tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015, 356, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The role of pyruvate kinase M2 in anticancer therapeutic treatments (Review). Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, J.; Guo, J.; Li, Y.; Lian, S.; Guo, W.; Yang, H.; Kong, F.; Zhen, L.; Guo, L.; et al. Progress in the biological function of alpha-enolase. Anim. Nutr. 2016, 2, 12–17. [Google Scholar] [CrossRef]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.-B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Hochachka, P.; Rupert, J.; Goldenberg, L.; Gleave, M.; Kozlowski, P. Going malignant: The hypoxia-cancer connection in the prostate. BioEssays 2002, 24, 749–757. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Sarrats, A.; Pérez-Bueno, F.; Oliveras, G.; Buxó, M.; Brunet, J.; Viñas, G.; Miquel, T.P. Fatty acid synthase expression and its association with clinico-histopathological features in triple-negative breast cancer. Oncotarget 2017, 8, 74391–74405. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Tan, W.; Fu, J.; Zhang, W. [Significance of fatty acid synthase expression in non-small cell lung cancer]. Zhonghua zhong liu za zhi Chin. J. Oncol. 2002, 24, 271–273. [Google Scholar]
- Agustsson, T.; Rydén, M.; Hoffstedt, J.; Van Harmelen, V.; Dicker, A.; Laurencikiene, J.; Isaksson, B.; Permert, J.; Arner, P. Mechanism of Increased Lipolysis in Cancer Cachexia. Cancer Res. 2007, 67, 5531–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Fan, X.-X.; He, J.; Pan, H.; Liyan, H.; Huang, L.; Jiang, Z.; Yao, X.-J.; Liu, L.; Leung, E.L.-H.; et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget 2016, 7, 39970–39979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osugi, J.; Yamaura, T.; Muto, S.; Okabe, N.; Matsumura, Y.; Hoshino, M.; Higuchi, M.; Suzuki, H.; Gotoh, M. Prognostic impact of the combination of glucose transporter 1 and ATP citrate lyase in node-negative patients with non-small lung cancer. Lung Cancer 2015, 88, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Hanai, J.-I.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein Kinase (MAPK) and Phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J. Cell. Physiol. 2011, 227, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Salvador, M.M.; de Cedrón, M.G.; Rubio, J.M.; Martínez, S.F.; Martínez, R.S.; Casado, E.; de Molina, A.R.; Sereno, M. Lipid metabolism and lung cancer. Crit. Rev. Oncol. 2017, 112, 31–40. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Liu, X.; Wang, H.; Li, J.; Dai, L.; Li, J.; Dong, C. Lung adenocarcinoma cell-derived exosomal miR-21 facilitates osteoclastogenesis. Gene 2018, 666, 116–122. [Google Scholar] [CrossRef]
- Khaidakov, M.; Mehta, J.L. Oxidized LDL Triggers Pro-Oncogenic Signaling in Human Breast Mammary Epithelial Cells Partly via Stimulation of MiR-21. PLoS ONE 2012, 7, e46973. [Google Scholar] [CrossRef]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Ni, K.; Wang, D.; Xu, H.; Mei, F.; Wu, C.; Liu, Z.; Zhou, B. miR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int. 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-D.; Feng, Q.-C.; Qi, Y.; Cui, G.; Zhao, S. PPARGC1A is upregulated and facilitates lung cancer metastasis. Exp. Cell Res. 2017, 359, 356–360. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Milne, J.L.S. Structure and Regulation of Pyruvate Dehydrogenases. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 321–328. [Google Scholar]
- Yang, C.; Wang, S.; Ruan, H.; Li, B.; Cheng, Z.; He, J.; Zuo, Q.; Yu, C.; Wang, H.; Lv, Y.; et al. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J. Cancer 2019, 10, 918–926. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Segawa, H.; Miyamoto, K.-I.; Uchino, H.; Takeda, E.; Endou, H. Expression Cloning and Characterization of a Transporter for Large Neutral Amino Acids Activated by the Heavy Chain of 4F2 Antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016, 26, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sah, J.F.; Beard, L.; Willson, J.K.V.; Markowitz, S.D.; Guda, K. The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosom. Cancer 2008, 47, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Griffiths, B.; Chung, Y.-L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wu, X.; Liu, B.; Wang, C.; Liu, Y.; Zhou, Q.; Xu, K. MiR-26a enhances metastasis potential of lung cancer cells via AKT pathway by targeting PTEN. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Li, N.; Zhou, X. Increased miR-21a provides metabolic advantages through suppression of FBP1 expression in non-small cell lung cancer cells. Am. J. Cancer Res. 2017, 7, 2121–2130. [Google Scholar]
- Lv, X.; Yao, L.; Zhang, J.; Han, P.; Li, C. Inhibition of microRNA-155 sensitizes lung cancer cells to irradiation via suppression of HK2-modulated glucose metabolism. Mol. Med. Rep. 2016, 14, 1332–1338. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Zhang, X.; Feng, X.; Fan, X.; Jin, Z. The crosstalk between microRNAs and the Wnt/β-catenin signaling pathway in cancer. Oncotarget 2016, 8, 14089–14106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-T.; Lee, S.Y.; Lim, S.H.; Yu, S.-L.; Ku, G.W.; Jeong, I.B.; Kwon, S.J.; Kim, J.H.; Kwon, S.J.; Kang, J.; et al. microRNA 181a-5p Reprogramed Glucose and Lipid Metabolism in Non- Small Cell Lung Cancer. J. Cancer Sci. Clin. Oncol. 2018, 5, 205. [Google Scholar]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Contractor, T.; Harris, C.R. p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.-C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 Broadly Influences Gene Expression and Promotes Apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.; Asselin, M.-C.; Reymen, B.; Jackson, A.; Lambin, P.; West, C.M.L.; O’Connor, J.P.B.; Faivre-Finn, C. Targeting Hypoxia to Improve Non–Small Cell Lung Cancer Outcome. J. Natl. Cancer Inst. 2017, 110, 14–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Mi, D.; Yang, K.; Cao, N.; Tian, J.; Li, Z.; Ma, B. The expression of hypoxia-inducible factor-1α and its clinical significance in lung cancer: A systematic review and meta-analysis. Swiss Med. Wkly. 2013, 143, w13855. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Linke, S.; Dias, J.M.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; Bilton, R.L.; et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.; et al. The Asparaginyl Hydroxylase Factor Inhibiting HIF-1α Is an Essential Regulator of Metabolism. Cell Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Cao, X.; Zhang, W.; Pan, G.; Yi, Q.; Zhong, W.; Yan, D. MicroRNA-31-5p enhances the Warburg effect via targeting FIH. FASEB J. 2019, 33, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, N. MicroRNA-21 modulates radiation resistance through upregulation of hypoxia-inducible factor-1α-promoted glycolysis in non-small cell lung cancer cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef]
- Byun, Y.; Choi, Y.-C.; Jeong, Y.; Lee, G.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, K. MiR-200c downregulates HIF-1α and inhibits migration of lung cancer cells. Cell. Mol. Biol. Lett. 2019, 24, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.-T.; Chen, P.-S.; Johansson, G.; Chu, C.-Y.; Wang, M.-Y.; Jeng, Y.-M.; Yu, S.-L.; Chen, J.-S.; Chang, K.-J.; Jee, S.-H.; et al. MicroRNA-519c Suppresses Hypoxia-Inducible Factor-1α Expression and Tumor Angiogenesis. Cancer Res. 2010, 70, 2675–2685. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Fortunato, O.; Boeri, M.; Verri, C.; Moro, M.; Sozzi, G. Therapeutic Use of MicroRNAs in Lung Cancer. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Van Rooij, E.; Olson, E.N. MicroRNAs: Powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Investig. 2007, 117, 2369–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.-H.; Ko, Y.H. Diagnostic and Therapeutic Implications of microRNAs in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2020, 21, 8782. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, J.R.; Wang, W.X.; Hébert, S.S.; Wilfred, B.R.; Mao, G.; Nelson, P.T. The miR-15/107 group of microRNA genes: Evolutionary biology, cellular functions, and roles in human diseases. J. Mol. Biol. 2010, 402, 491–509. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Crawford, M.; Mao, Y.; Lee, R.J.; Davis, I.C.; Elton, T.S.; Lee, L.J.; Nana-Sinkam, S.P. Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol. Ther. Nucleic Acids 2013, 2, e84. [Google Scholar] [CrossRef]
- Rai, K.; Takigawa, N.; Ito, S.; Kashihara, H.; Ichihara, E.; Yasuda, T.; Shimizu, K.; Tanimoto, M.; Kiura, K. Liposomal Delivery of MicroRNA-7–Expressing Plasmid Overcomes Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistance in Lung Cancer Cells. Mol. Cancer Ther. 2011, 10, 1720–1727. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a Lung Cancer Therapeutic Based on the Tumor Suppressor MicroRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, M.A.; Valdecanas, D.; Zhang, X.; Zhan, Y.; Bhardwaj, V.; Calin, G.A.; Komaki, R.; Giri, D.K.; Quini, C.C.; Wolfe, T.; et al. Therapeutic Delivery of miR-200c Enhances Radiosensitivity in Lung Cancer. Mol. Ther. 2014, 22, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; De La Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic Delivery of Tumor Suppressor microRNA Mimics Using a Neutral Lipid Emulsion Inhibits Lung Tumors in Mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Barger, J.F.; Nana-Sinkam, S.P. MicroRNA as tools and therapeutics in lung cancer. Respir. Med. 2015, 109, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Soriano, A.; Jubierre, L.; Almazán-Moga, A.; Molist, C.; Roma, J.; de Toledo, J.S.; Gallego, S.; Segura, M.F. microRNAs as pharmacological targets in cancer. Pharmacol. Res. 2013, 75, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setién, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Gitt, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Kauppinen, S.; Lund, A.H. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.F.R.; Rebhan, M.A.E.; Crivelli, S.E.M.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014, 42, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; Van Der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Shimura, E.; Nagasawa, A.; Aiuchi, T.; Suda, Y.; Hamada, Y.; Ikegami, D.; Iwasawa, C.; Arakawa, K.; Igarashi, K.; et al. Chronic treatment of non-small-cell lung cancer cells with gefitinib leads to an epigenetic loss of epithelial properties associated with reductions in microRNA-155 and -200c. PLoS ONE 2017, 12, e0172115. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ding, C.-M.; Li, Y.-X.; Peng, J.-C.; Geng, N.; Qin, W.-W. Over-regulation of microRNA-133b inhibits cell proliferation of cisplatin-induced non-small cell lung cancer cells through PI3K/Akt and JAK2/STAT3 signaling pathway by targeting EGFR. Oncol. Rep. 2018, 39, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-X.; Yue, Z.; Wang, P.-Y.; Li, Y.-J.; Xin, J.-X.; Pang, M.; Zheng, Q.-Y.; Xie, S.-Y. Cisplatin upregulates MSH2 expression by reducing miR-21 to inhibit A549 cell growth. Biomed. Pharmacother. 2013, 67, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Qiao, F.; Wang, Y.; Xu, Y.; Shang, Y. Curcumin inhibits cell proliferation and induces apoptosis of human non-small cell lung cancer cells through the upregulation of miR-192-5p and suppression of PI3K/Akt signaling pathway. Oncol. Rep. 2015, 34, 2782–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.-L.; Zhou, S.-J.; Zhang, X.-M.; Chen, H.-Q.; Liu, W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed. Pharmacother. 2016, 78, 74–80. [Google Scholar] [CrossRef]
- Yagishita, S.; Fujita, Y.; Kitazono, S.; Ko, R.; Nakadate, Y.; Sawada, T.; Kitamura, Y.; Shimoyama, T.; Maeda, Y.; Takahashi, F.; et al. Chemotherapy-Regulated microRNA-125–HER2 Pathway as a Novel Therapeutic Target for Trastuzumab-Mediated Cellular Cytotoxicity in Small Cell Lung Cancer. Mol. Cancer Ther. 2015, 14, 1414–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, C.; Li, Y.; Wang, P.; Yue, Z.; Xie, S. miR-98 regulates cisplatin-induced A549 cell death by inhibiting TP53 pathway. Biomed. Pharmacother. 2011, 65, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shen, H.; Guo, R.; Sun, J.; Gao, W.; Shu, Y. Combine therapy of gefitinib and fulvestrant enhances antitumor effects on NSCLC cell lines with acquired resistance to gefitinib. Biomed. Pharmacother. 2012, 66, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-H.; Wang, X.; Feng, Q. EGCG enhances the efficacy of cisplatin by downregulating HSA-miR-98-5p in NSCLC A549 cells. Nutr. Cancer. 2014, 66, 636–644. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Metabolic/Signaling Pathway | MiRNA | Up/Downregulated | Target | Function(s) | Reference |
|---|---|---|---|---|---|
| Glucose metabolism | miR-144 | Downregulated | GLUT1 | Enhances Warburg effect | [17] |
| miR-124 | Downregulated | GLUT1, HK2, p-Akt1/2 | Enhances Warburg effect and ATP generation | [18] | |
| miR-143 | Downregulated | HK2 | Enhances Warburg effect | [19] | |
| miR-206 | Downregulated | HK2 | Enhances Warburg effect | [20] | |
| miR-133 | Downregulated | PKM2 | Enhances Warburg effect and radiation resistance | [21] | |
| miR-29 | Downregulated | ENO1, PGAM1, PGK1, CMPK1 | Facilitates glycolysis | [22] | |
| Lipid metabolism | miR-33b | Downregulated | LDHA | Enhances aerobic glycolysis and tumor invasion | [23] |
| miR-21 | Upregulated | HBP1, CD36 | Promotes proliferation and increase intracellular lipid influx | [24,25] | |
| miR-182 | Upregulated | PDK4/PDH axis | Enhances de novo lipogenesis and JNK activation | [26] | |
| Amino acid metabolism | miR-126 | Downregulated | SLC7A5 | Facilitates amino acid exchange and activate mTOR | [27] |
| PI3K/Akt/PTEN | miR-21, miR-155 | Upregulated | SOCS1, SOCS6, PTEN | PTEN inhibition | [24] |
| miR-125 | Upregulated | p-Akt, GSK3β | PI3K/Akt and GSK3β/Wnt/β-catenin activation | [28] | |
| c-Myc | miR-451 | Downregulated | c-Myc, GSK3β | Enhanced metabolic dysregulation and migration | [29] |
| Hypoxia | miR-34 | Upregulation | SIRT1 | Facilitate mutant p53 expression via a feedback loop | [30,31] |
| miR-199-5p | Activates HIF-1α | [32] | |||
| miR-210 | Upregulated | NDUFA4, SDHD | Mitochondria disfiguration and cristae formation | [33] | |
| miR-31-5p | Upregulated | FIH | Promotes hypoxia and enhances aerobic glycolysis | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azizi, M.I.H.N.; Othman, I.; Naidu, R. The Role of MicroRNAs in Lung Cancer Metabolism. Cancers 2021, 13, 1716. https://doi.org/10.3390/cancers13071716
Azizi MIHN, Othman I, Naidu R. The Role of MicroRNAs in Lung Cancer Metabolism. Cancers. 2021; 13(7):1716. https://doi.org/10.3390/cancers13071716
Chicago/Turabian StyleAzizi, Mohamed Iman Hidayat Nor, Iekhsan Othman, and Rakesh Naidu. 2021. "The Role of MicroRNAs in Lung Cancer Metabolism" Cancers 13, no. 7: 1716. https://doi.org/10.3390/cancers13071716