
ImmunoAIzer: A Deep Learning-Based Computational Framework to Characterize Cell Distribution and Gene Mutation in Tumor Microenvironment

 ,
, 
Abstract
:Simple Summary
Abstract
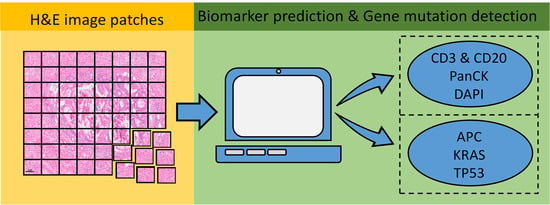
1. Introduction
2. Materials and Methods
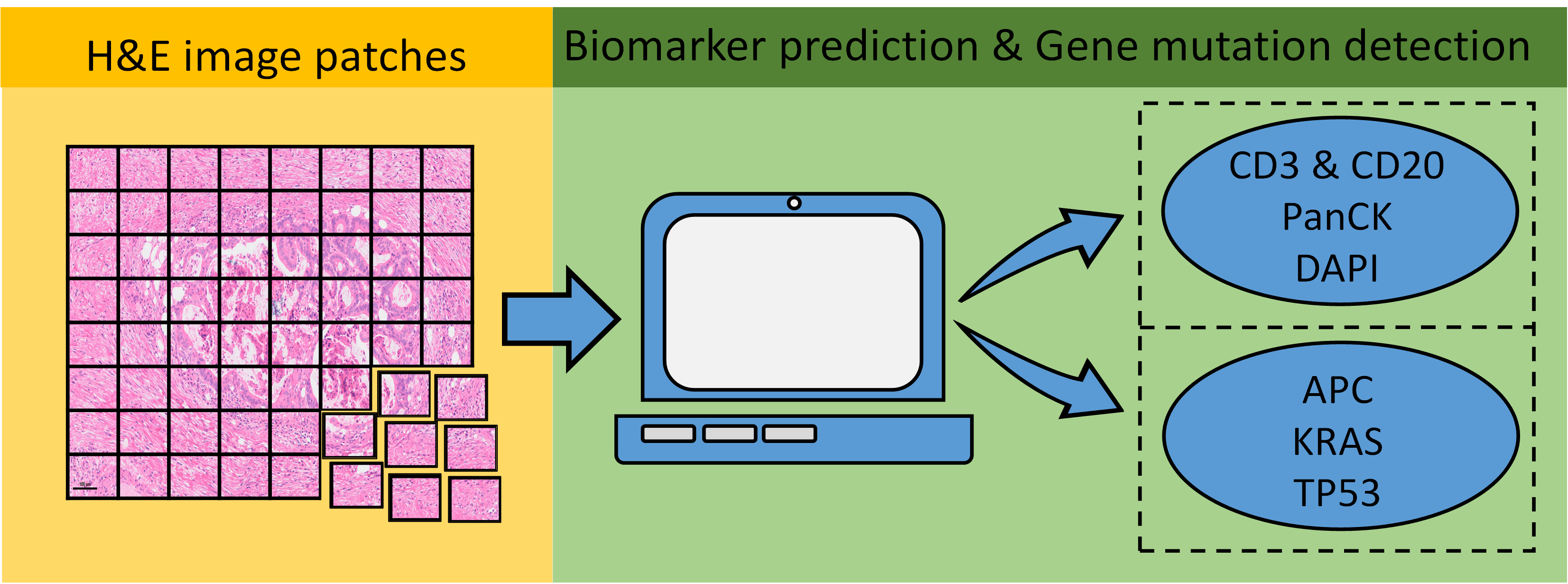
2.1. Dataset Establishment
2.2. CBDPN Training and Validation
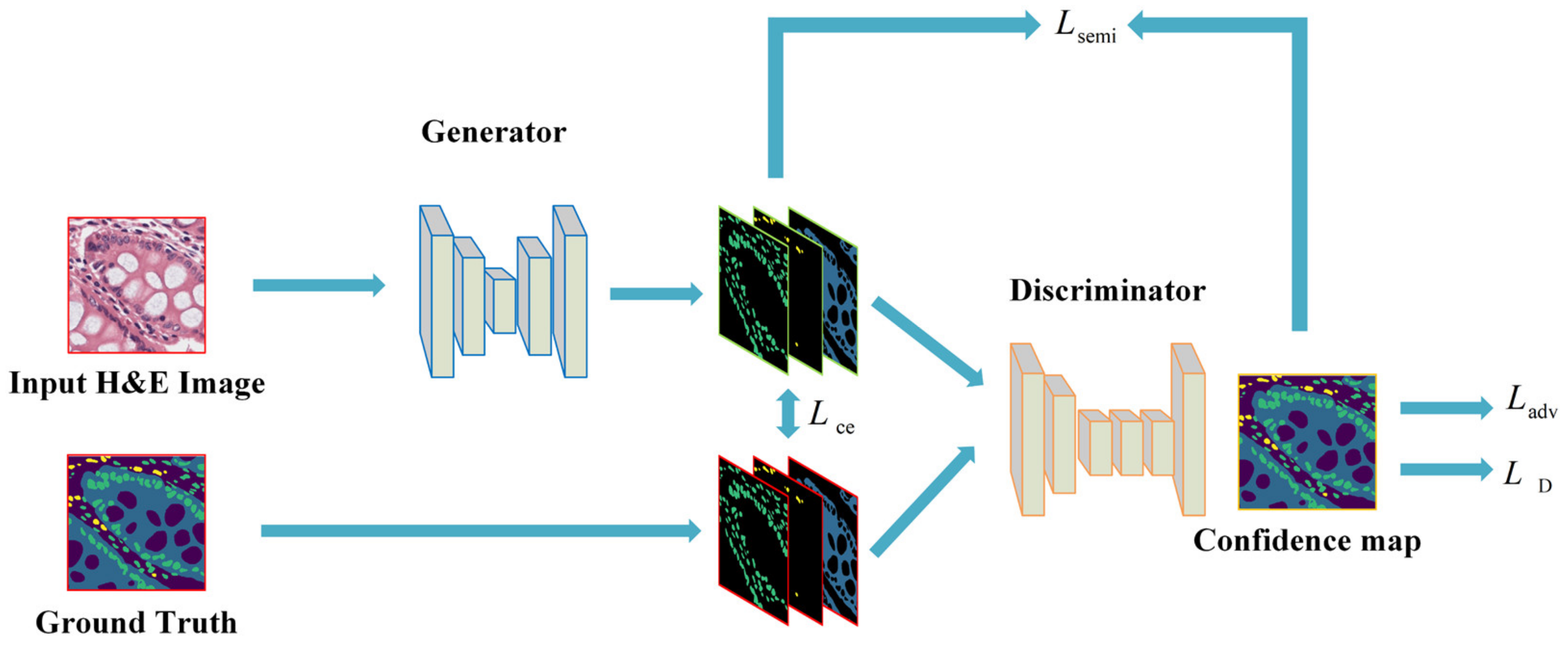
2.2.1. Semi-Supervised Mechanism
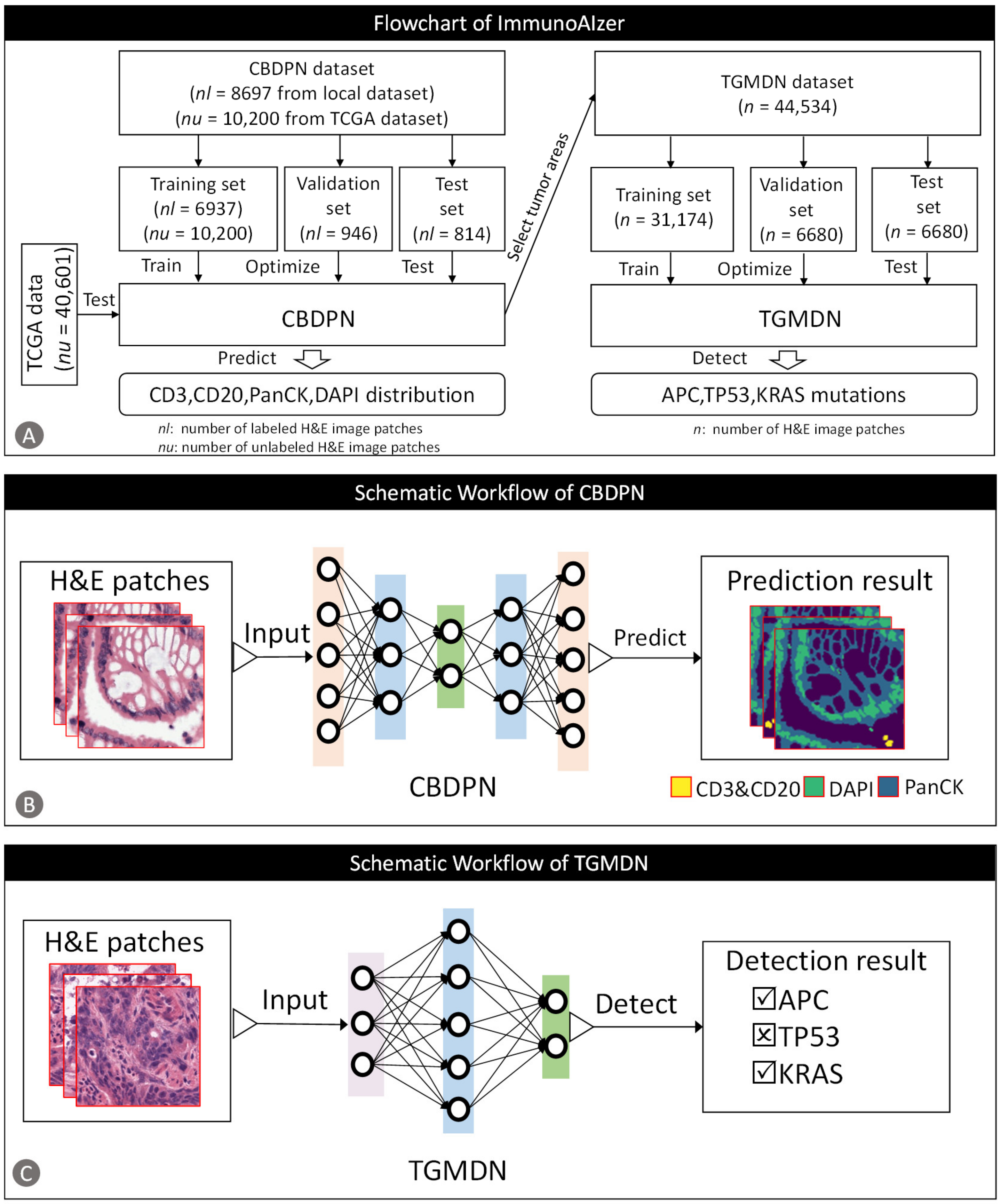
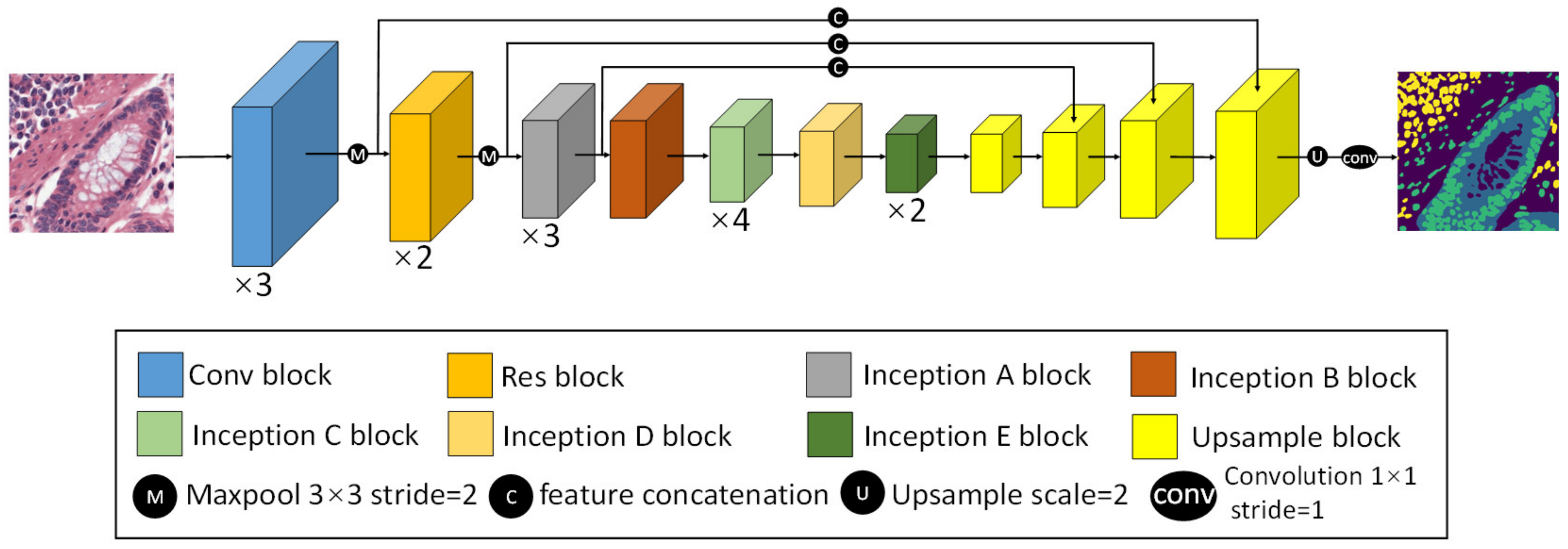
2.2.2. CBPDN Structure
2.2.3. Test Set Validation
2.2.4. TCGA Dataset Validation
2.2.5. CBDPN-Based Cell Quantification
2.3. TGMDN Training and Validation
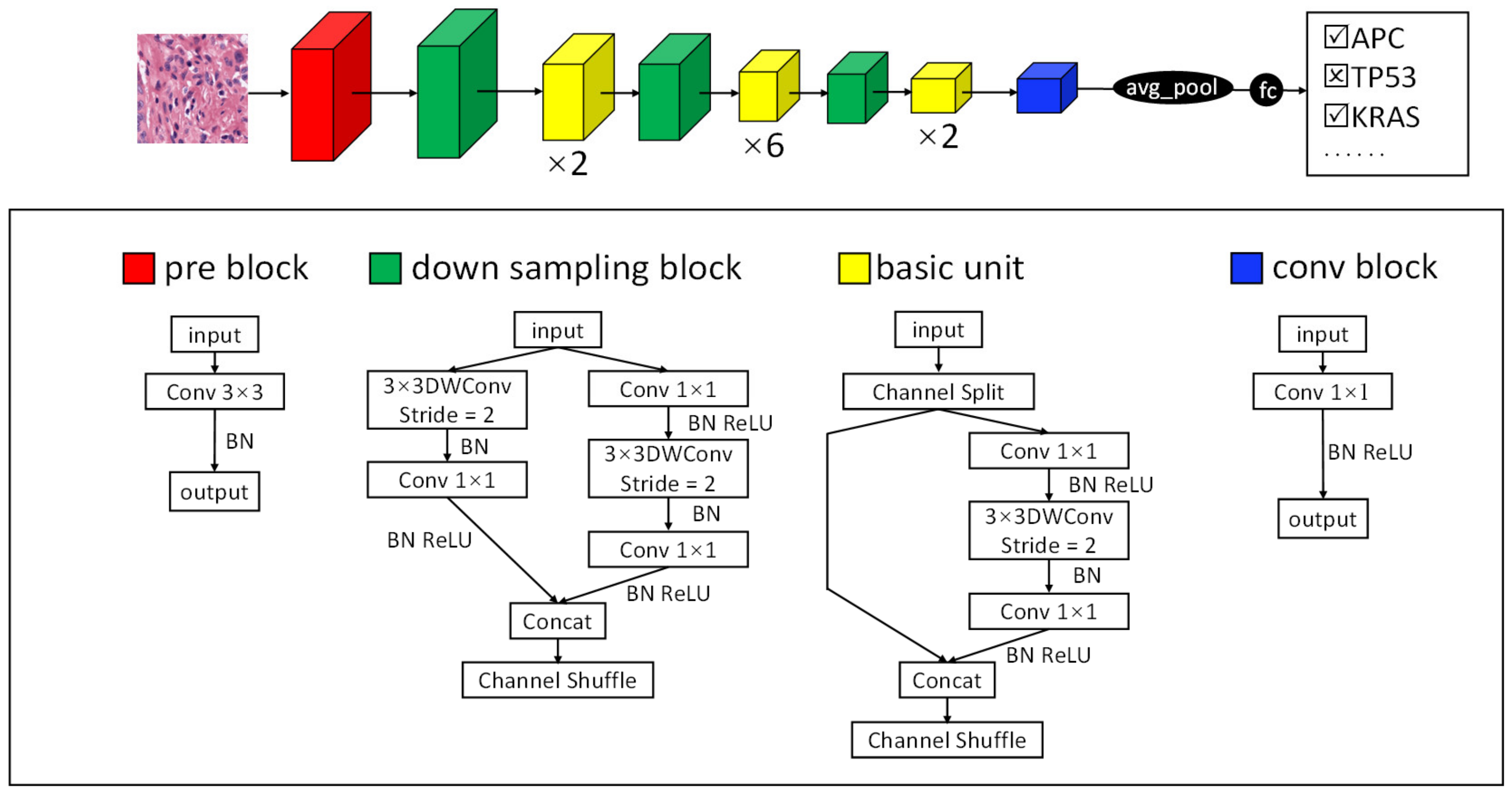
2.3.1. TGMDN Structure
2.3.2. TGMDN Evaluation
2.4. Implementation Details
3. Results
3.1. CBDPN for the Prediction of Cellular Biomarker Distribution in TME
3.1.1. Fully-Supervised Experiment Results
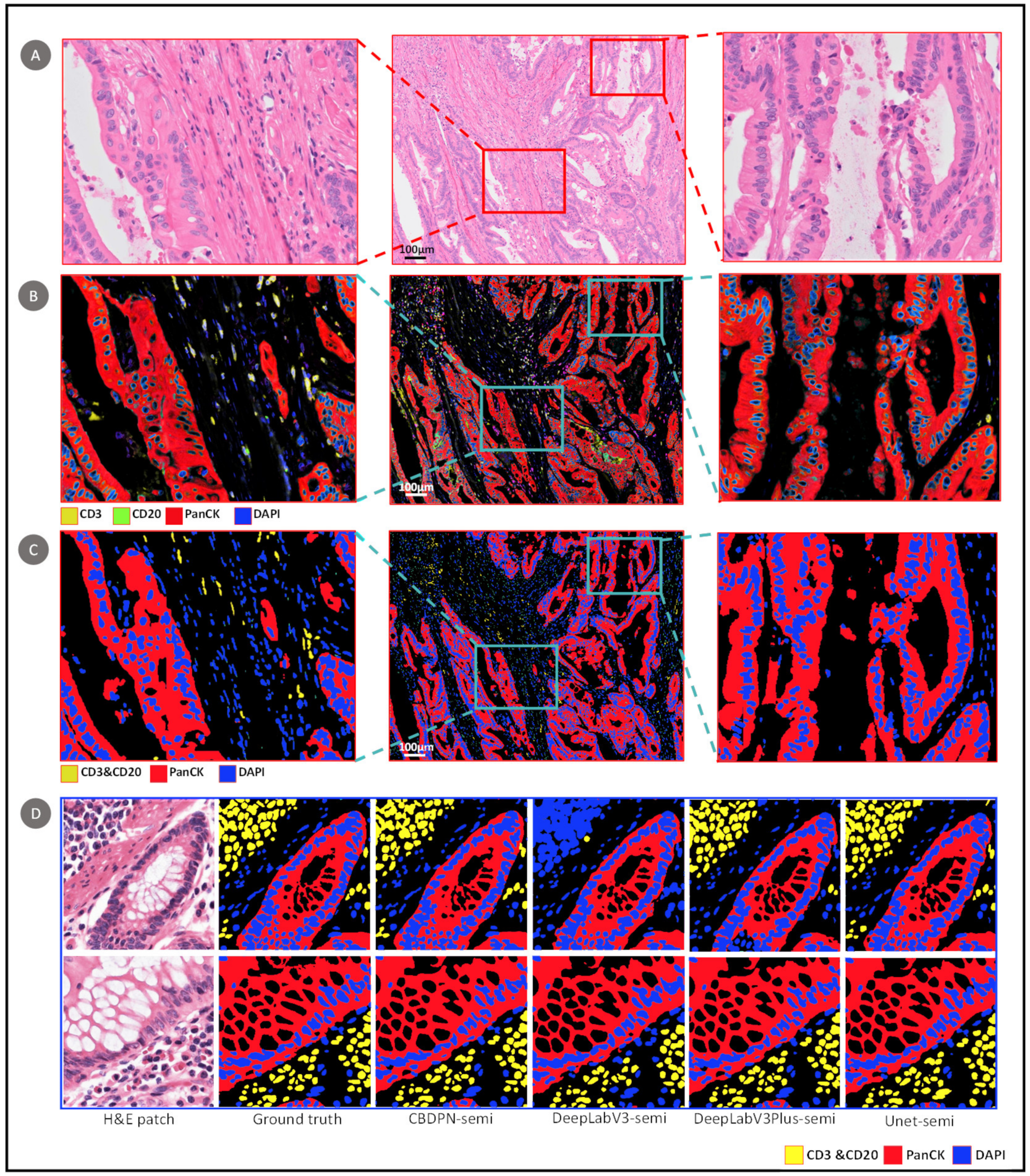
3.1.2. Semi-Supervised Experiment Results
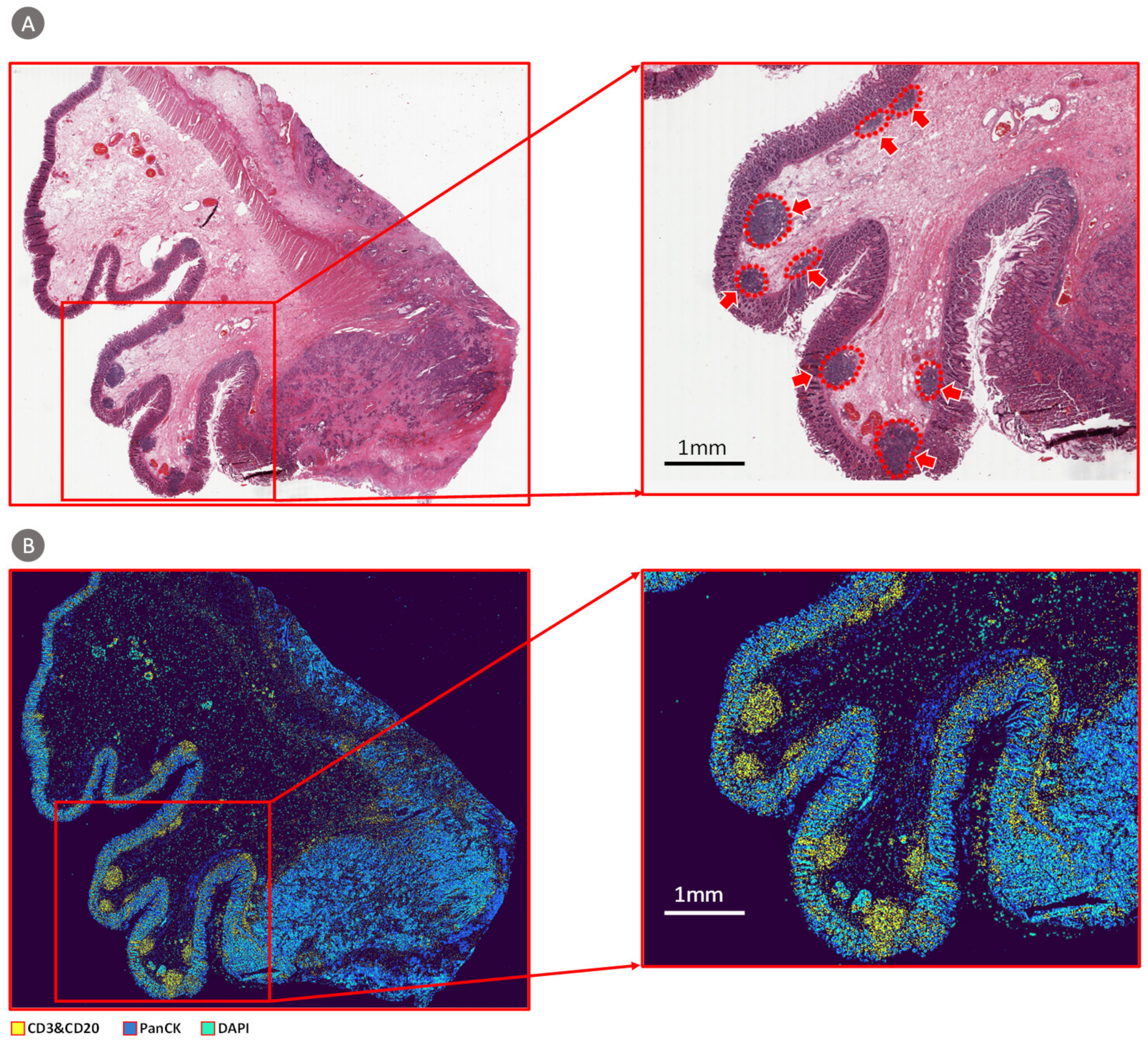
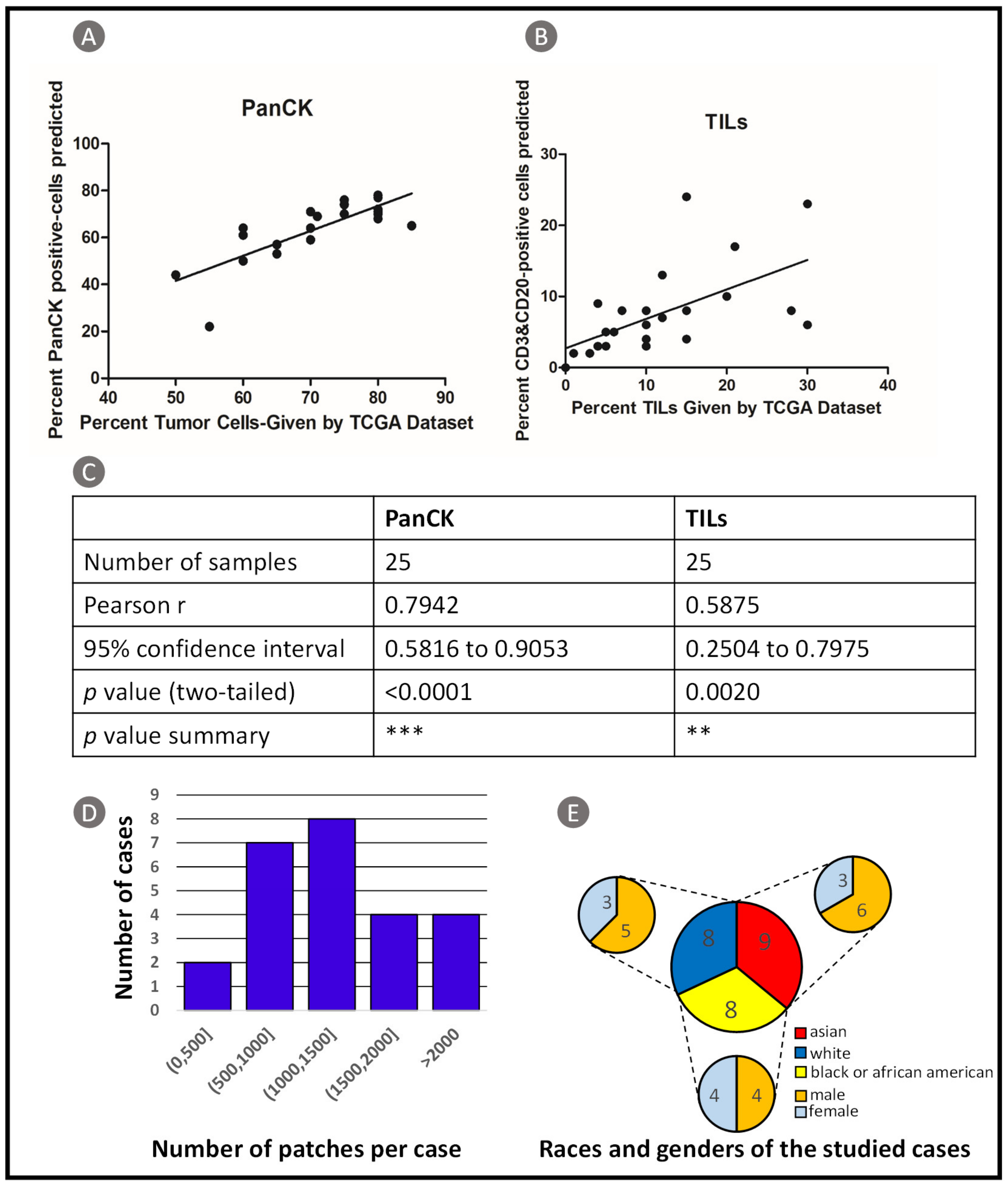
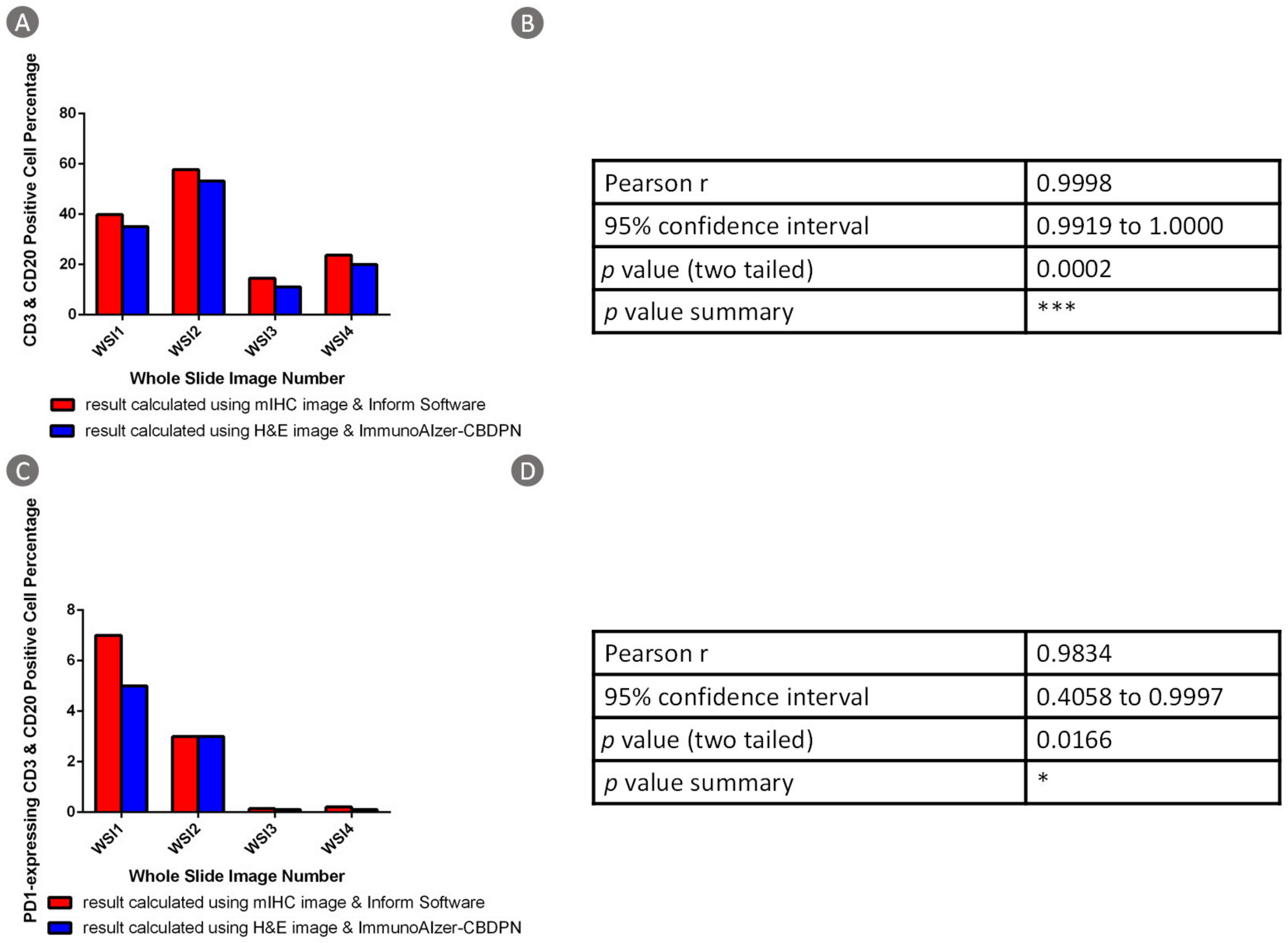
3.1.3. TCGA Dataset Validation Results
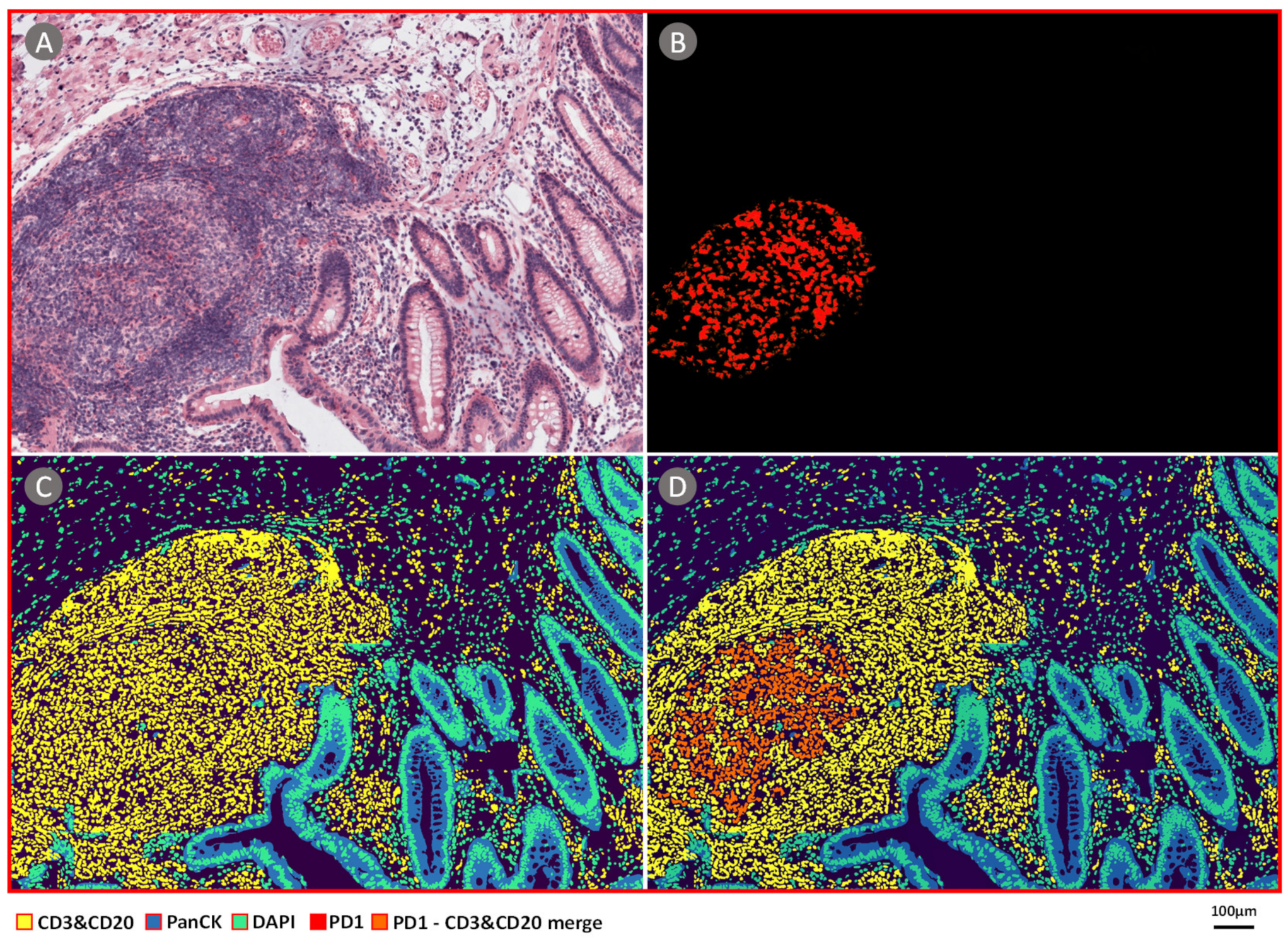
3.1.4. CBDPN-Based Cell Quantification Analysis
3.2. TGMDN for the Detection of Tumor Gene Mutations
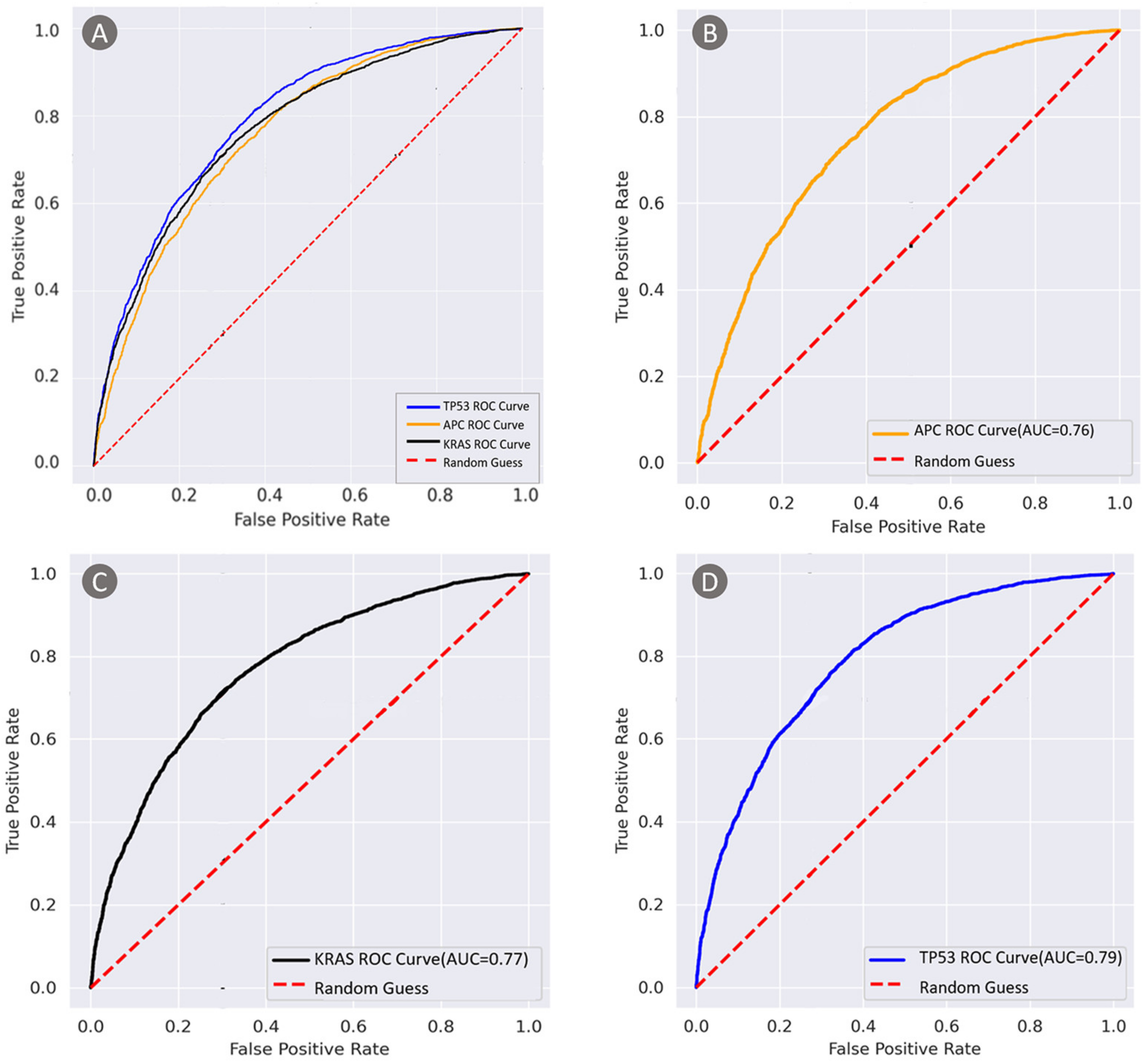
3.2.1. Detection of Tumor Gene Mutations from H&E Images
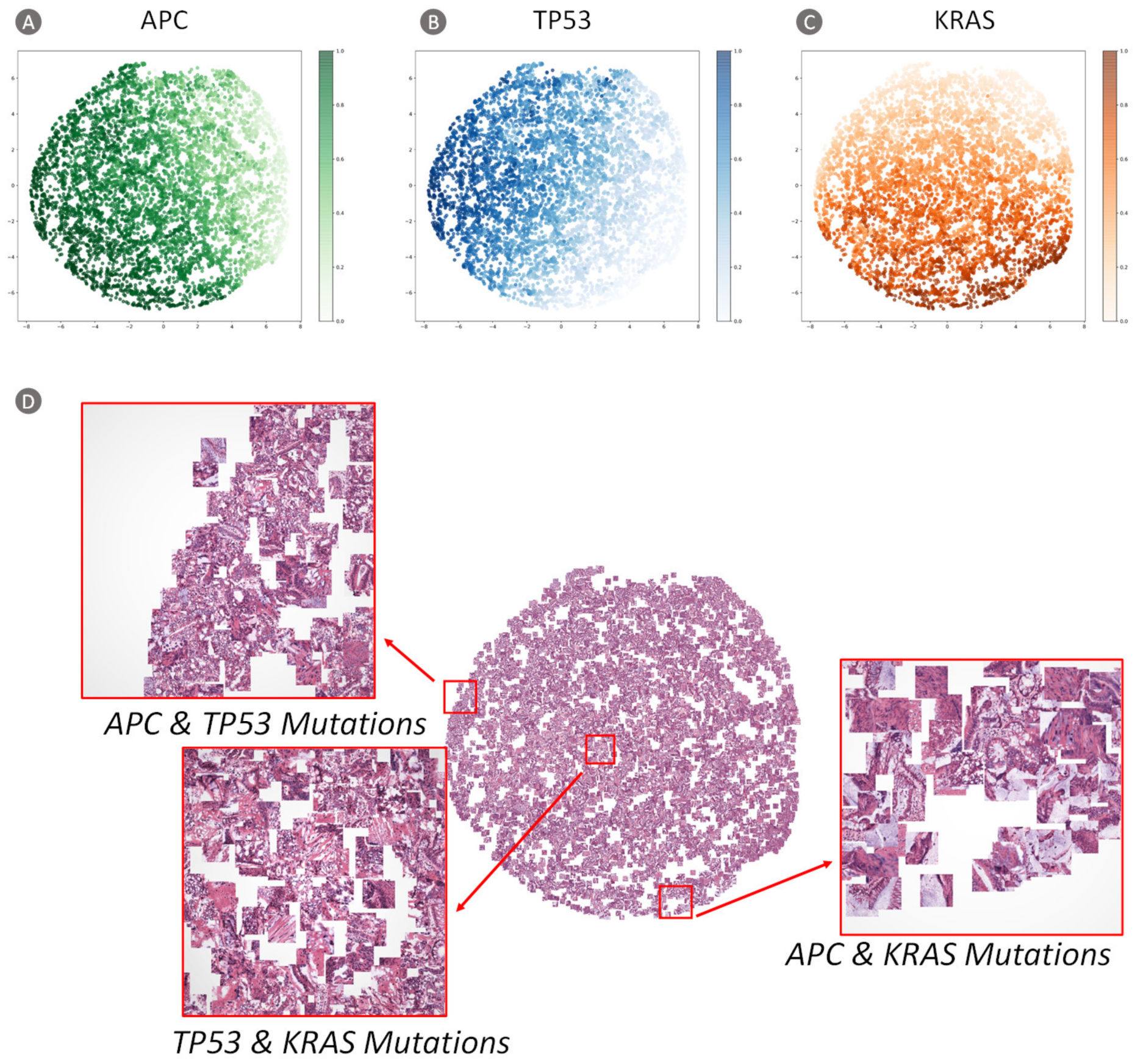
3.2.2. Visualization of Network Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
- Coarse block matching: Generally, the original WSIs are extremely large in size and thus difficult to process in their entirety owing to RAM limitations. To address this problem, we randomly selected 15 candidate blocks (~5000 × ~4000 pixels) in each mIHC staining WSI. Then, a normalized correlation matrix was calculated by correlating each of the ~5000 × ~4000-pixel blocks with the corresponding block extracted from the whole-slide grayscale H&E image of the same size. The block with the highest correlation score was considered to be the coarsely matched H&E block for the two staining blocks.
- Global registration: After acquiring coarsely matched block pairs. A global registration step was carried out to correct the slight rotation angle. We extracted feature vectors (descriptors) and their corresponding locations from the block pairs; we then matched the features using the descriptors [46]. Next, the M-estimator sample consensus algorithm was used to calculate the transformation matrix [47]. After the rotation was applied, the images were cropped, removing 50 pixels on each side to eliminate the undefined areas that resulted from the rotation.
- Elastic registration: After Step 2, an elastic registration between H&E image blocks and globally registered mIHC image blocks was conducted by applying a diffeomorphic demons algorithm [48] to correct the distortions induced by warping and various aberrations.
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.J.O. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.C.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Jin, Y.H.; Sun, W.; Fang, J.J.; Zheng, J.J.; Tian, J. Advances in molecular imaging of immune checkpoint targets in malignancies: Current and future prospect. Eur. Radiol. 2019, 29, 4294–4302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Qi, Y.F.; Jin, Z.G.; Tian, J. Noninvasive imaging in cancer immunotherapy: The way to precision medicine. Cancer Lett. 2019, 466, 13–22. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A New Approach To the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Roth, A.D.; Delorenzi, M.; Tejpar, S.; Yan, P.; Klingbiel, D.; Fiocca, R.; d’Ario, G.; Cisar, L.; Labianca, R.; Cunningham, D.; et al. Integrated Analysis of Molecular and Clinical Prognostic Factors in Stage II/III Colon Cancer. J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Dawson, D.M.; Willis, J.; Willson, J.K.V. Focus on colon cancer. Cancer Cell 2002, 1, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Westra, J.L.; Schaapveld, M.; Hollema, H.; de Boer, J.P.; Kraak, M.M.J.; de Jong, D.; ter Elst, A.; Mulder, N.H.; Buys, C.H.C.M.; Hofstra, R.M.W.; et al. Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J. Clin. Oncol. 2005, 23, 5635–5643. [Google Scholar] [CrossRef]
- Liao, W.T.; Overman, M.J.; Boutin, A.T.; Shang, X.Y.; Zhao, D.; Dey, P.; Li, J.X.; Wang, G.C.; Lan, Z.D.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Kalra, J.; Baker, J. Multiplex Immunohistochemistry for Mapping the Tumor Microenvironment. In Signal Transduction Immunohistochemistry: Methods and Protocols; Kalyuzhny, A.E., Ed.; Springer: New York, NY, USA, 2017; pp. 237–251. [Google Scholar] [CrossRef]
- Stack, E.C.; Wang, C.C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef]
- Blom, S.; Paavolainen, L.; Bychkov, D.; Turkki, R.; Maki-Teeri, P.; Hemmes, A.; Valimaki, K.; Lundin, J.; Kallioniemi, O.; Pellinen, T. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci. Rep. 2017, 7, 15580. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, G.V.; Fergie, M.; Berks, M.; Harkness, E.F.; Hulleman, J.; Brentnall, A.R.; Cuzick, J.; Evans, D.G.; Astley, S.M. Prediction of reader estimates of mammographic density using convolutional neural networks. J. Med. Imaging 2019, 6, 031405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanella, G.; Hanna, M.G.; Geneslaw, L.; Miraflor, A.; Silva, V.W.K.; Busam, K.J.; Brogi, E.; Reuter, V.E.; Klimstra, D.S.; Fuchs, T.J. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 2019, 25, 1301–1309. [Google Scholar] [CrossRef]
- Rivenson, Y.; Wang, H.D.; Wei, Z.S.; de Haan, K.; Zhang, Y.B.; Wu, Y.C.; Gunaydin, H.; Zuckerman, J.E.; Chong, T.; Sisk, A.E.; et al. Virtual histological staining of unlabelled tissue-autofluorescence images via deep learning. Nat. Biomed. Eng. 2019, 3, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Saltz, J.; Gupta, R.; Hou, L.; Kurc, T.; Singh, P.; Nguyen, V.; Samaras, D.; Shroyer, K.R.; Zhao, T.H.; Batiste, R.; et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep. 2018, 23, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, E.M.; Yang, S.J.; Ando, D.M.; Javaherian, A.; Skibinski, G.; Lipnick, S.; Mount, E.; O’Neil, A.; Shah, K.; Lee, A.K.; et al. In Silico Labeling: Predicting Fluorescent Labels in Unlabeled Images. Cell 2018, 173, 792–803. [Google Scholar] [CrossRef] [Green Version]
- Burlingame, E.A.; Margolin, A.A.; Gray, J.W.; Chang, Y.H. SHIFT: Speedy histopathological-to-immunofluorescent translation of whole slide images using conditional generative adversarial networks. In Medical Imaging 2018: Digital Pathology; SPIE: Houston, TX, USA, 2018; Volume 10581, p. 1058105. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A.J.N.m. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Litjens, G.; Kooi, T.; Bejnordi, B.E.; Setio, A.A.A.; Ciompi, F.; Ghafoorian, M.; van der Laak, J.A.W.M.; van Ginneken, B.; Sanchez, C.I. A survey on deep learning in medical image analysis. Med. Image Anal. 2017, 42, 60–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapelle, O.; Scholkopf, B.; Zien, A. Semi-supervised learning (chapelle, o. et al., eds.; 2006) [book reviews]. IEEE Trans. Neural Netw. 2009, 20, 542. [Google Scholar] [CrossRef]
- Brown, J.R.; Wimberly, H.; Lannin, D.R.; Nixon, C.; Rimm, D.L.; Bossuyt, V. Multiplexed Quantitative Analysis of CD3, CD8, and CD20 Predicts Response to Neoadjuvant Chemotherapy in Breast Cancer. Clin. Cancer Res. 2014, 20, 5995–6005. [Google Scholar] [CrossRef] [Green Version]
- Macenko, M.; Niethammer, M.; Marron, J.S.; Borland, D.; Woosley, J.T.; Guan, X.J.; Schmitt, C.; Thomas, N.E. A Method for Normalizing Histology Slides for Quantitative Analysis. In Proceedings of the 2009 IEEE International Symposium on Biomedical Imaging: From Nano To Macro, Boston, MA, USA, 28 June–1 July 2009; Volume 1–2, pp. 1107–1110. [Google Scholar] [CrossRef]
- Vahadane, A.; Peng, T.Y.; Sethi, A.; Albarqouni, S.; Wang, L.C.; Baust, M.; Steiger, K.; Schlitter, A.M.; Esposito, I.; Navab, N. Structure-Preserving Color Normalization and Sparse Stain Separation for Histological Images. IEEE T Med. Imaging 2016, 35, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.C.; Tsai, Y.H.; Liou, Y.T.; Lin, Y.Y.; Yang, M.H. Adversarial Learning for Semi-Supervised Semantic Segmentation. In Proceedings of the British Machine Vision Conference, Newcastle, UK, 3–6 September 2018. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Szegedy, C.; Vanhoucke, V.; Ioffe, S.; Shlens, J.; Wojna, Z. Rethinking the Inception Architecture for Computer Vision. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 26 June–1 July 2016. [Google Scholar] [CrossRef] [Green Version]
- Ronneberger, O.; Fischer, P.; Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Munich, Germany, 5–9 October 2015; Volume 9351, pp. 234–241. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-C.; Papandreou, G.; Schroff, F.; Adam, H. Rethinking Atrous Convolution for Semantic Image Segmentation. arXiv 2017, arXiv:1706.05587. [Google Scholar]
- Chen, L.-C.; Zhu, Y.; Papandreou, G.; Schroff, F.; Adam, H. Encoder-decoder with atrous separable convolution for semantic image segmentation. In Proceedings of the European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 801–818. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Wyss, J.; Dislich, B.; Koelzer, V.H.; Galvan, J.A.; Dawson, H.; Hadrich, M.; Inderbitzin, D.; Lugli, A.; Zlobec, I.; Berger, M.D. Stromal PD-1/PD-L1 Expression Predicts Outcome in Colon Cancer Patients. Clin. Colorectal Cancer 2019, 18, E20–E38. [Google Scholar] [CrossRef]
- Sehdev, A.; Cramer, H.M.; Ibrahim, A.A.; Younger, A.E.; O’Neil, B.H. Pathological Complete Response with Anti-PD-1 Therapy in a Patient with Microsatellite Instable High, BRAF Mutant Metastatic Colon Cancer: A Case Report and Review of Literature. Discov. Med. 2016, 21, 341–347. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.S.; Latham, B.; Diepeveen, L.A.; Mou, L.J.; Laurent, G.J.; Elsegood, C.; Ochoa-Callejero, L.; Yeoh, G.C. InForm software: A semi-automated research tool to identify presumptive human hepatic progenitor cells, and other histological features of pathological significance. Sci. Rep. 2018, 8, 3418. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Zhang, X.; Zheng, H.-T.; Sun, J. ShuffleNet V2: Practical Guidelines for Efficient CNN Architecture Design. In Proceedings of the European Conference on Computer Vision, Munich, Germany, 8–14 September 2018; pp. 122–138. [Google Scholar]
- Kather, J.N.; Heij, L.R.; Grabsch, H.I.; Loeffler, C.; Echle, A.; Muti, H.S.; Krause, J.; Niehues, J.M.; Sommer, K.A.; Bankhead, P.J.N.C. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 2020, 1, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2625. [Google Scholar]
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic Differentiation in Pytorch. Available online: https://openreview.net/forum?id=BJJsrmfCZ (accessed on 20 February 2021).
- Gurcan, M.N.; Boucheron, L.E.; Can, A.; Madabhushi, A.; Rajpoot, N.M.; Yener, B. Histopathological image analysis: A review. IEEE Rev. Biomed. Eng. 2009, 2, 147–171. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.C.; Yang, Y.; Wang, J.; Xu, W.; Yuille, A.L. Attention to Scale: Scale-aware Semantic Image Segmentation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 26 June–1 July 2016; pp. 3640–3649. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.G. Distinctive image features from scale-invariant keypoints. Int. J. Comput. Vis. 2004, 60, 91–110. [Google Scholar] [CrossRef]
- Torr, P.H.S.; Zisserman, A. MLESAC: A new robust estimator with application to estimating image geometry. Comput. Vis. Image Underst. 2000, 78, 138–156. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, T.; Pennec, X.; Perchant, A.; Ayache, N. Diffeomorphic demons: Efficient non-parametric image registration. Neuroimage 2009, 45, S61–S72. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network | Accuracy | Precision | Recall | Dice | IoU |
|---|---|---|---|---|---|
| UNet | 0.873 | 0.799 | 0.871 | 0.825 | 0.714 |
| DeepLab V3 | 0.856 | 0.776 | 0.836 | 0.797 | 0.677 |
| DeepLab V3+ | 0.863 | 0.778 | 0.858 | 0.805 | 0.690 |
| CBPDN | 0.875 | 0.806 | 0.865 | 0.827 | 0.717 |
| Network | Accuracy | Precision | Recall | Dice | IoU |
|---|---|---|---|---|---|
| UNet | 0.891 | 0.804 | 0.863 | 0.824 | 0.711 |
| DeepLab V3 | 0.868 | 0.789 | 0.859 | 0.813 | 0.701 |
| DeepLab V3+ | 0.872 | 0.803 | 0.865 | 0.823 | 0.712 |
| CBPDN | 0.904 | 0.854 | 0.901 | 0.872 | 0.788 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, C.; Wang, Y.; Lu, Z.; An, Y.; Wang, H.; Kong, L.; Du, Y.; Tian, J. ImmunoAIzer: A Deep Learning-Based Computational Framework to Characterize Cell Distribution and Gene Mutation in Tumor Microenvironment. Cancers 2021, 13, 1659. https://doi.org/10.3390/cancers13071659
Bian C, Wang Y, Lu Z, An Y, Wang H, Kong L, Du Y, Tian J. ImmunoAIzer: A Deep Learning-Based Computational Framework to Characterize Cell Distribution and Gene Mutation in Tumor Microenvironment. Cancers. 2021; 13(7):1659. https://doi.org/10.3390/cancers13071659
Chicago/Turabian StyleBian, Chang, Yu Wang, Zhihao Lu, Yu An, Hanfan Wang, Lingxin Kong, Yang Du, and Jie Tian. 2021. "ImmunoAIzer: A Deep Learning-Based Computational Framework to Characterize Cell Distribution and Gene Mutation in Tumor Microenvironment" Cancers 13, no. 7: 1659. https://doi.org/10.3390/cancers13071659