
Overexpression of Human Syndecan-1 Protects against the Diethylnitrosamine-Induced Hepatocarcinogenesis in Mice

 , , , ,
, , , ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Human Syndecan-1-Transgenic (hSDC+/+) Mice
2.2. Hepatocarcinogenesis
2.3. ELISA
2.4. Phospho-Receptor Tyrosine Kinase (pRTK) Array
2.5. Phosphorylation Antibody Array Analysis
2.6. Western Blot
2.7. Immunohistochemistry
2.8. Proteomics, Bioinformatics
2.8.1. Surface Digestion of FFPE Tissues
2.8.2. Chromatography and Mass Spectrometry
2.8.3. Protein Identification and Label-Free Quantitation
2.8.4. Protein Interaction Analysis
2.9. Statistical Analysis
3. Results
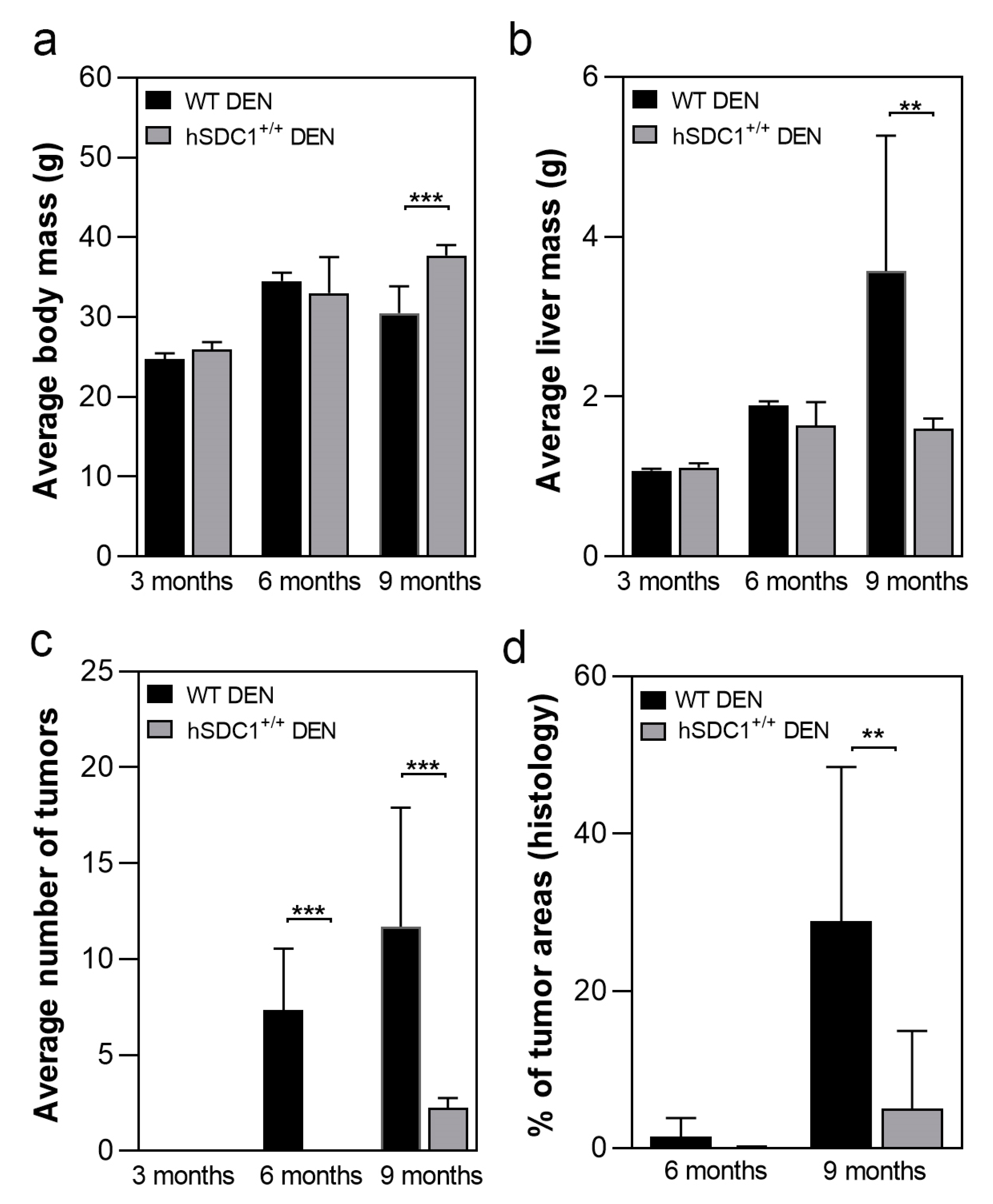
3.1. The Development of DEN-Induced Liver Tumors Was Delayed in hSDC1+/+ Mice
3.2. Expression of Mouse and Human Syndecan-1 in the Livers of WT and hSDC1+/+ Mice
3.3. Shedding of Mouse and Human Syndecan-1
3.4. pRTK Array Indicates Downregulation of Receptor Activation in DEN-Treated hSDC1+/+ Mice
3.5. Different Early Phase Response to DEN Exposure in the Lipid Metabolism of WT vs. hSDC+/+ Mice
3.6. TGF-β1 Expression in Control and DEN-Exposed Livers
3.7. Downstream Signaling Effects of the Overexpression of hSDC1
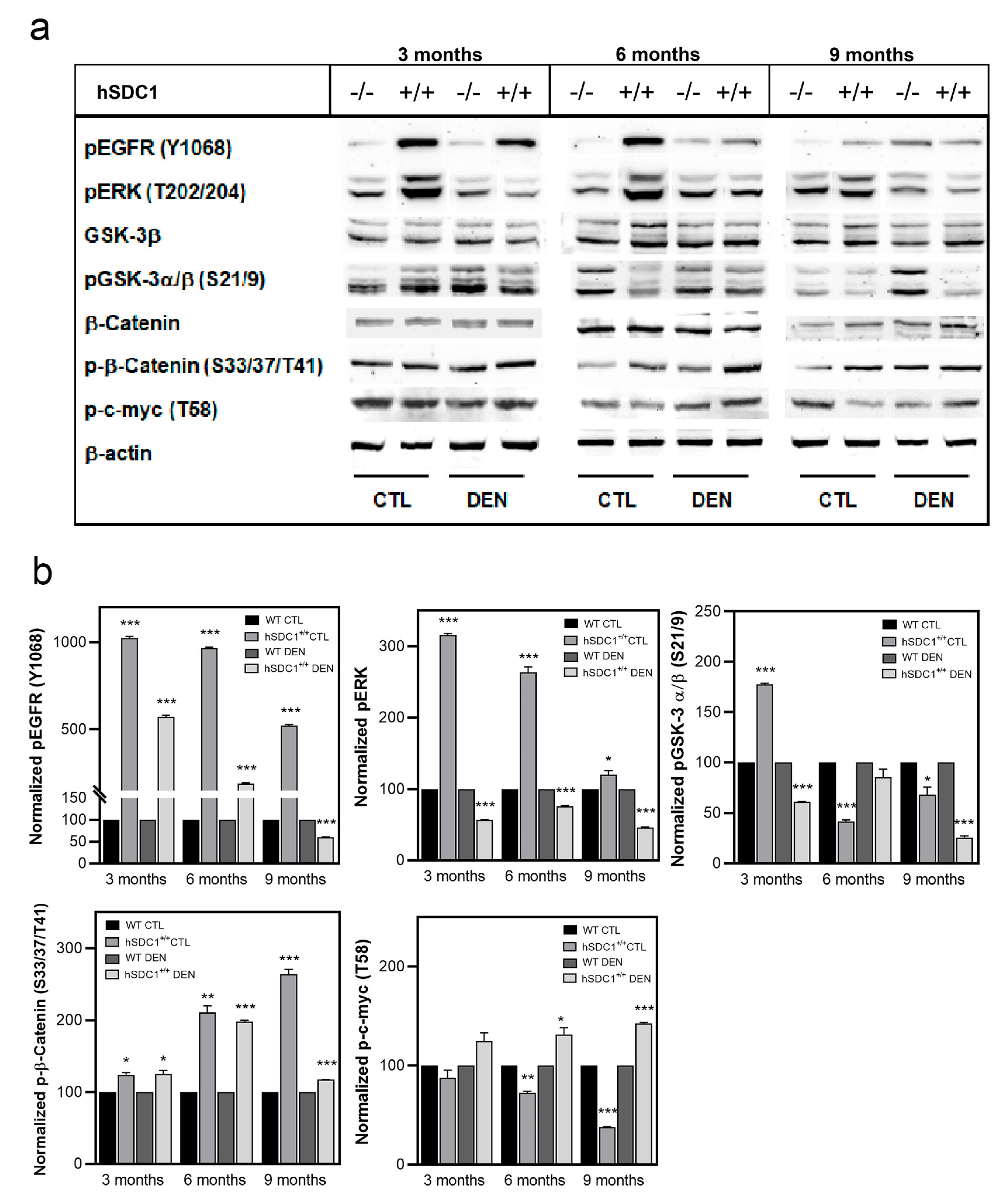
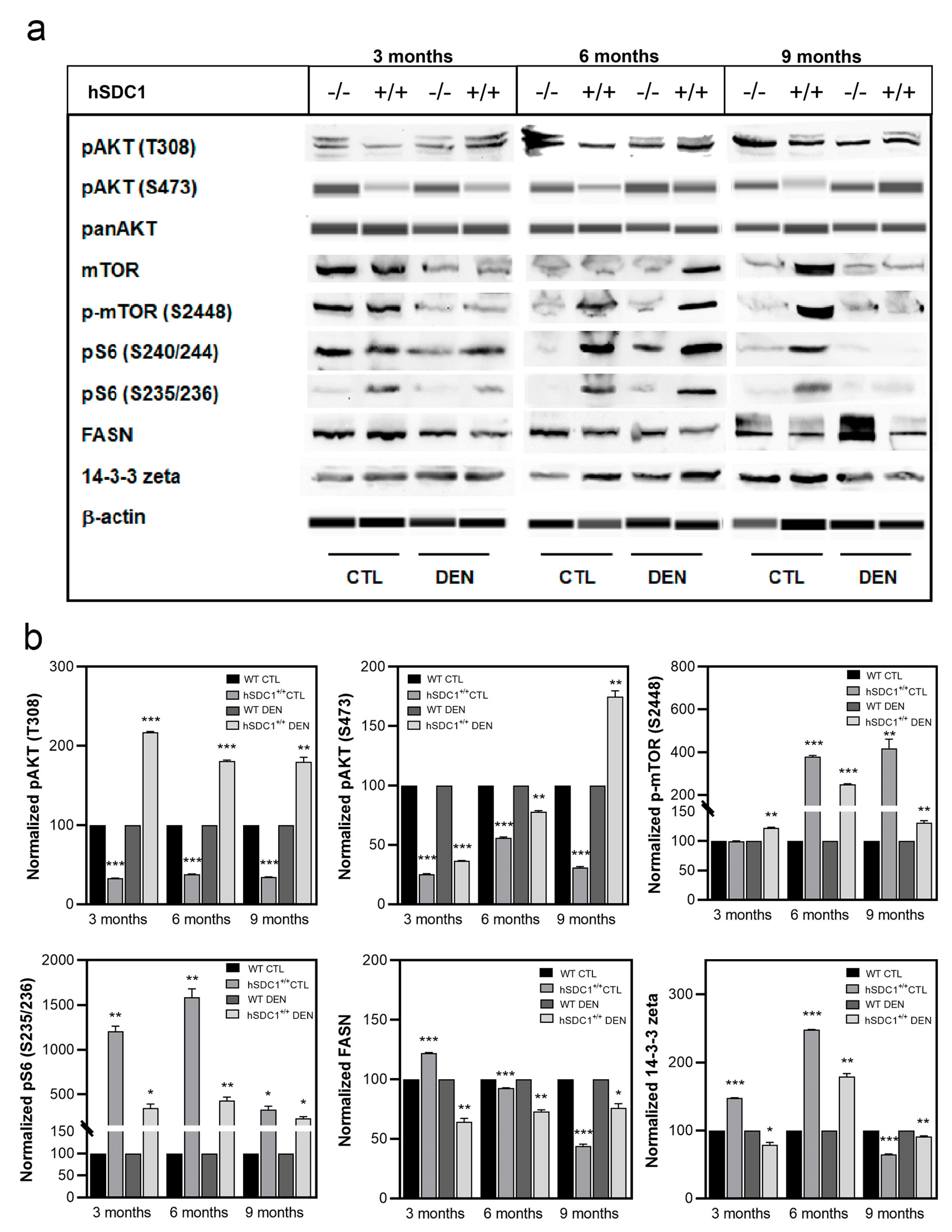
3.7.1. Western Blots Indicate Attenuated Pro-Proliferatory Signals in hSDC1+/+ DEN
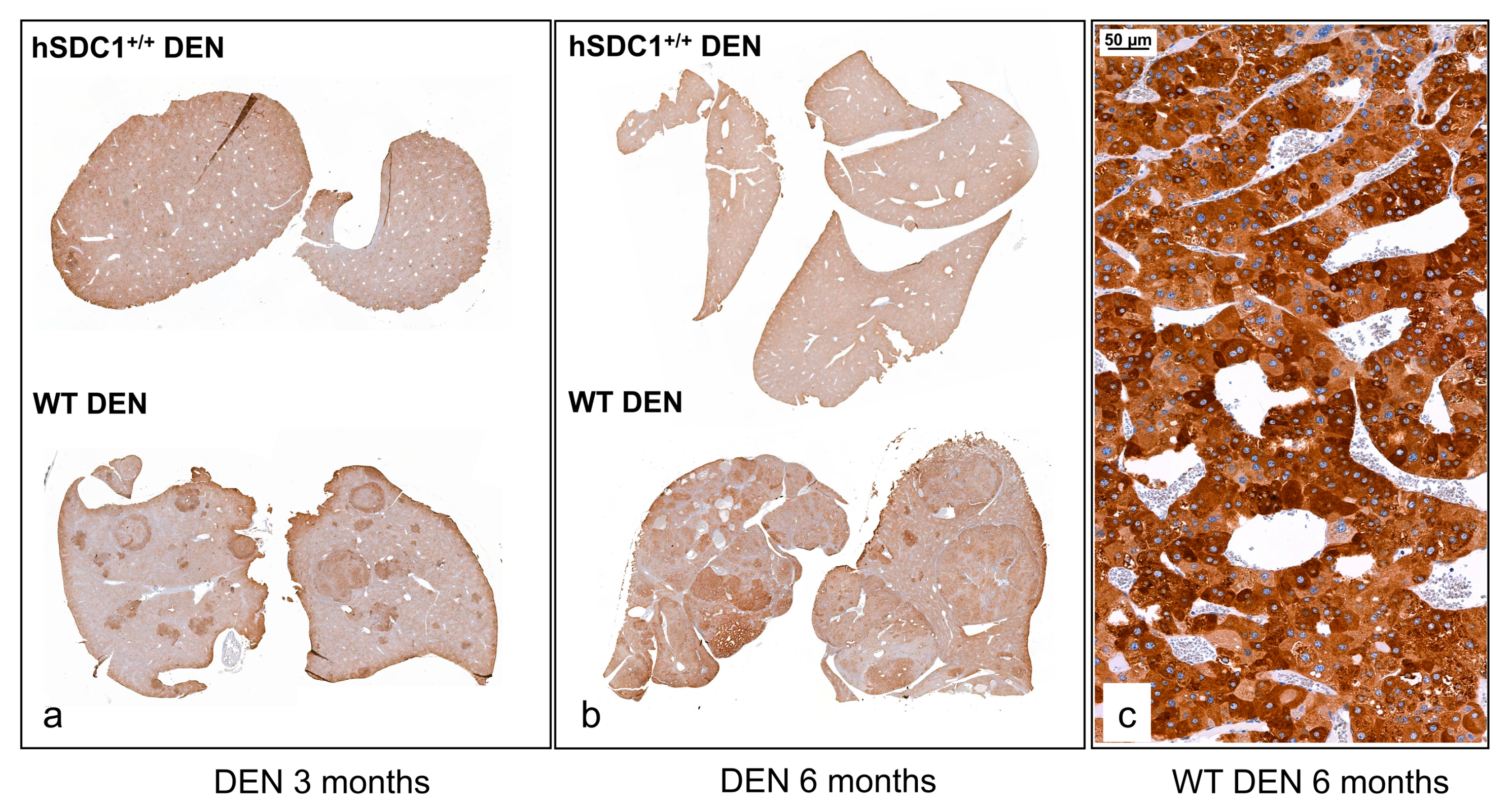
3.7.2. Immunohistochemistry Reveals Further Differences between WT DEN and hSDC1+/+ DEN Livers at Month 3
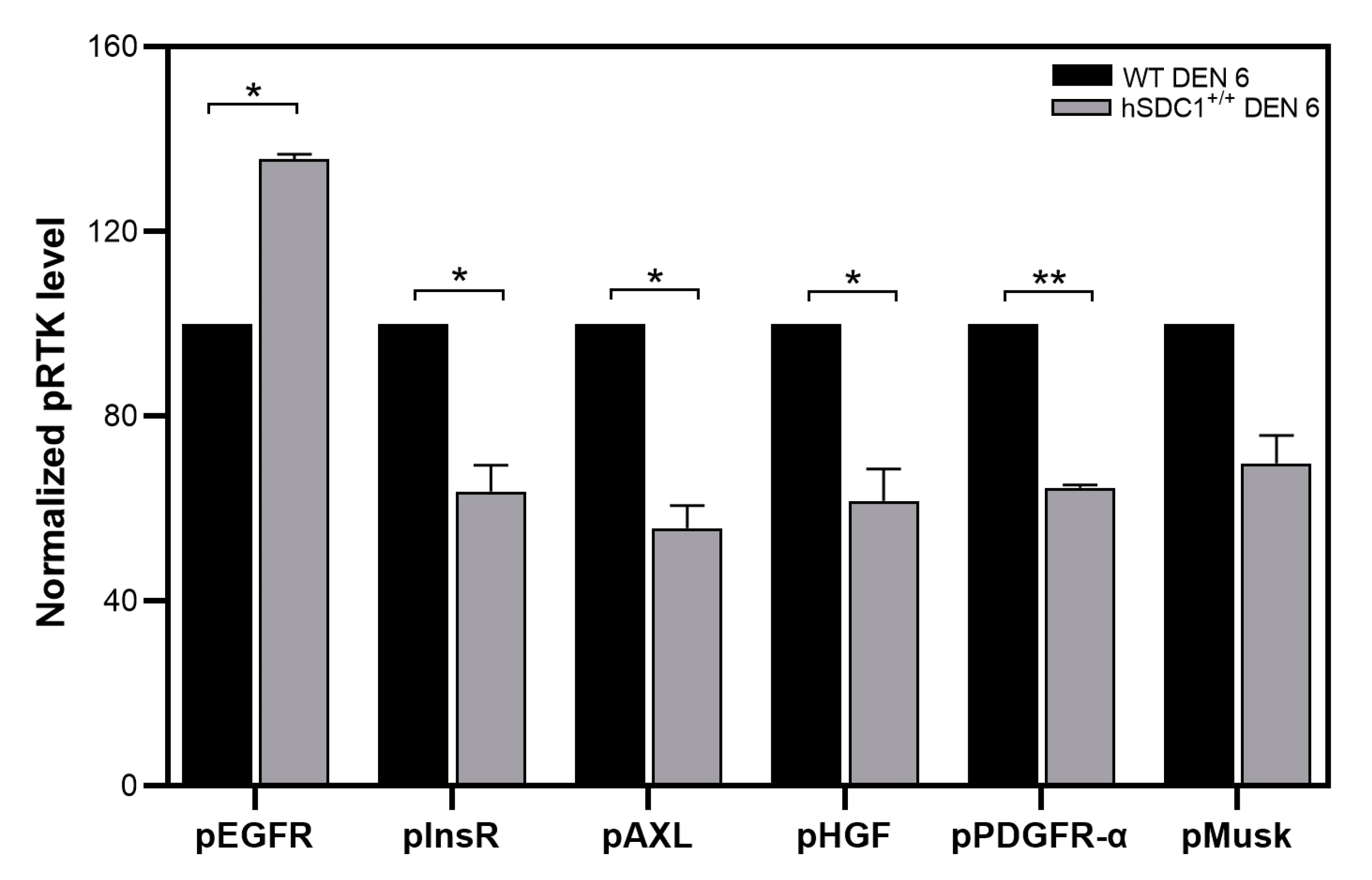
3.7.3. Phospho-Kinase Array at Month 6 Confirms Suppressed Cancer Progression in hSDC1+/+ DEN
3.8. Proteomics Analysis and STRING Interaction Networks
3.8.1. Untreated (WT CTL and hSDC1+/+ CTL) Livers
3.8.2. Alteration in DEN-Exposed Liver at Month 3
3.8.3. Alterations in DEN-Exposed Livers at Month 6
3.8.4. Alterations in DEN-Exposed Livers at Month 9
4. Discussion
4.1. Hepatocarcinogenesis
4.2. Changes in Syndecan-1 Shedding and Receptor Tyrosine Kinase Activity
4.3. Alterations in Lipid Metabolism
4.4. Changes in the Levels of TGF-β1
4.5. Hepatocarcinogenesis Modified in hSDC1+/+ Mice is Characterized by the Downregulation of β-Catenin and mTOR Pathways
4.5.1. Downregulation of the β-Catenin Pathway
4.5.2. Downregulation of the mTOR Pathway and Associated Changes in Intermediary Metabolism
4.6. Changes in Cell Cycle Regulation
4.7. Proteomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambaerts, K.; Van Dyck, S.; Mortier, E.; Ivarsson, Y.; Degeest, G.; Luyten, A.; Vermeiren, E.; Peers, B.; David, G.; Zimmermann, P. Syntenin, a syndecan adaptor and an Arf6 phosphatidylinositol 4,5-bisphosphate effector, is essential for epiboly and gastrulation cell movements in zebrafish. J. Cell Sci. 2012, 125, 1129–1140. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Montmasson, M.; Terradot, L.; Rousselle, P. Syndecans as Cell Surface Receptors in Cancer Biology. A Focus on their Interaction with PDZ Domain Proteins. Front. Pharm. 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couchman, J.R.; Gopal, S.; Lim, H.C.; Nørgaard, S.; Multhaupt, H.A. Fell-Muir Lecture: Syndecans: From peripheral coreceptors to mainstream regulators of cell behaviour. Int. J. Exp. Pathol 2015, 96, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Beauvais, D.M.; Rapraeger, A.C. Syndecans in tumor cell adhesion and signaling. Reprod. Biol. Endocrinol. 2004, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, S.; Søgaard, P.; Multhaupt, H.A.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Singh, A.; Shrinet, J.; Parniewska, M.M.; Fuxe, J.; Dobra, K.; Hjerpe, A. Mapping the Interactome of the Nuclear Heparan Sulfate Proteoglycan Syndecan-1 in Mesothelioma Cells. Biomolecules 2020, 10, 1034. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ramani, V.C.; Sanderson, R.D. Shed syndecan-1 translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: A novel mechanism of tumor-host cross-talk. J. Biol. Chem. 2015, 290, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Kovalszky, I.; Dudás, J.; Oláh-Nagy, J.; Pogány, G.; Töváry, J.; Timár, J.; Kopper, L.; Jeney, A.; Iozzo, R.V. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol. Cell Biochem. 1998, 183, 11–23. [Google Scholar] [CrossRef]
- Karászi, K.; Vigh, R.; Máthé, M.; Fullár, A.; Oláh, L.; Füle, T.; Papp, Z.; Kovalszky, I. Aberrant Expression of Syndecan-1 in Cervical Cancers. Pathol. Oncol. Res. 2020, 26, 2255–2264. [Google Scholar] [CrossRef]
- Szatmári, T.; Ötvös, R.; Hjerpe, A.; Dobra, K. Syndecan-1 in Cancer: Implications for Cell Signaling, Differentiation, and Prognostication. Dis. Markers 2015, 2015, 796052. [Google Scholar] [CrossRef] [Green Version]
- Stanford, K.I.; Bishop, J.R.; Foley, E.M.; Gonzales, J.C.; Niesman, I.R.; Witztum, J.L.; Esko, J.D. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Investig. 2009, 119, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Pruett, P.S.; Thompson, C.A.; DeLucas, L.D.; Sanderson, R.D. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J. Biol. Chem. 2012, 287, 9952–9961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regős, E.; Karászi, K.; Reszegi, A.; Kiss, A.; Schaff, Z.; Baghy, K.; Kovalszky, I. Syndecan-1 in Liver Diseases. Pathol. Oncol. Res. 2020, 26, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Hollósi, P.; Váncza, L.; Karászi, K.; Dobos, K.; Péterfia, B.; Tátrai, E.; Tátrai, P.; Szarvas, T.; Paku, S.; Szilák, L.; et al. Syndecan-1 Promotes Hepatocyte-Like Differentiation of Hepatoma Cells Targeting Ets-1 and AP-1. Biomolecules 2020, 10, 1356. [Google Scholar] [CrossRef]
- Regős, E.; Abdelfattah, H.H.; Reszegi, A.; Szilák, L.; Werling, K.; Szabó, G.; Kiss, A.; Schaff, Z.; Kovalszky, I.; Baghy, K. Syndecan-1 inhibits early stages of liver fibrogenesis by interfering with TGFβ1 action and upregulating MMP14. Matrix Biol. 2018, 68–69, 474–489. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turiák, L.; Shao, C.; Meng, L.; Khatri, K.; Leymarie, N.; Wang, Q.; Pantazopoulos, H.; Leon, D.R.; Zaia, J. Workflow for combined proteomics and glycomics profiling from histological tissues. Anal. Chem. 2014, 86, 9670–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turiák, L.; Ozohanics, O.; Tóth, G.; Ács, A.; Révész, Á.; Vékey, K.; Telekes, A.; Drahos, L. High sensitivity proteomics of prostate cancer tissue microarrays to discriminate between healthy and cancerous tissue. J. Proteom. 2019, 197, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Dudás, J.; Ramadori, G.; Knittel, T.; Neubauer, K.; Raddatz, D.; Egedy, K.; Kovalszky, I. Effect of heparin and liver heparan sulphate on interaction of HepG2-derived transcription factors and their cis-acting elements: Altered potential of hepatocellular carcinoma heparan sulphate. Biochem. J. 2000, 350 Pt 1, 245–251. [Google Scholar] [CrossRef]
- Lendorf, M.E.; Manon-Jensen, T.; Kronqvist, P.; Multhaupt, H.A.; Couchman, J.R. Syndecan-1 and syndecan-4 are independent indicators in breast carcinoma. J. Histochem. Cytochem. 2011, 59, 615–629. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Li, N.; Liao, F. Identification of Key Genes and Pathways in Pancreatic Cancer Gene Expression Profile by Integrative Analysis. Genes 2019, 10, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demoulin, J.B.; Ericsson, J.; Kallin, A.; Rorsman, C.; Rönnstrand, L.; Heldin, C.H. Platelet-derived growth factor stimulates membrane lipid synthesis through activation of phosphatidylinositol 3-kinase and sterol regulatory element-binding proteins. J. Biol. Chem. 2004, 279, 35392–35402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chait, A.; Ross, R.; Albers, J.J.; Bierman, E.L. Platelet-derived growth factor stimulates activity of low density lipoprotein receptors. Proc. Natl. Acad. Sci. USA 1980, 77, 4084–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, M.; Klimentidis, Y.C.; Casazza, K.; Chambers, M.M.; Cho, R.; Harbison, S.T.; Jumbo-Lucioni, P.; Zhang, S.; Leips, J.; Fernandez, J.R. A conserved role for syndecan family members in the regulation of whole-body energy metabolism. PLoS ONE 2010, 5, e11286. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zuo, D.; Chen, Y.; Li, W.; Liu, R.; He, Y.; Ren, L.; Zhou, L.; Deng, T.; Wang, X.; et al. Shed Syndecan-1 is involved in chemotherapy resistance via the EGFR pathway in colorectal cancer. Br. J. Cancer 2014, 111, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.M.; Esko, J.D. Hepatic heparan sulfate proteoglycans and endocytic clearance of triglyceride-rich lipoproteins. Prog. Mol. Biol. Transl. Sci. 2010, 93, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.I.; et al. Syndecan-1 is required to maintain intradermal fat and prevent cold stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.; Pilo, M.G.; Cigliano, A.; Latte, G.; Simile, M.M.; Ribback, S.; Dombrowski, F.; Evert, M.; Chen, X.; Calvisi, D.F. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle Georget. Tex. 2017, 16, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 20484. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.K.; Singh, N.; Sahu, D.K. TGF-β Mediated Crosstalk Between Malignant Hepatocyte and Tumor Microenvironment in Hepatocellular Carcinoma. Cancer Growth Metastasis 2014, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef]
- Pataki, C.A.; Couchman, J.R.; Brábek, J. Wnt Signaling Cascades and the Roles of Syndecan Proteoglycans. J. Histochem. Cytochem. 2015, 63, 465–480. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, V.C.; Markworth, J.F.; Cameron-Smith, D. Considerations on mTOR regulation at serine 2448: Implications for muscle metabolism studies. Cell Mol. Life Sci. 2017, 74, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- Pullen, N.; Thomas, G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997, 410, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, A. Akt is more than just a Bad kinase. Nature 1999, 401, 33–34. [Google Scholar] [CrossRef]
- Shang, N.; Arteaga, M.; Zaidi, A.; Stauffer, J.; Cotler, S.J.; Zeleznik-Le, N.J.; Zhang, J.; Qiu, W. FAK is required for c-Met/β-catenin-driven hepatocarcinogenesis. Hepatology 2015, 61, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gan, L.; Pan, H.; Guo, S.; He, X.; Olson, S.T.; Mesecar, A.; Adam, S.; Unterman, T.G. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 2002, 277, 45276–45284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiramongkol, Y.; Lam, E.W. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Invest 2006, 116, 2464–2472. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Chauhan, A.S.; Zhuang, L.; Gan, B. FoxO transcription factors in cancer metabolism. Semin. Cancer Biol. 2018, 50, 65–76. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, R.; Martinez, A.; Døskeland, S.O.; Haavik, J. The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 2011, 22, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Cui, L.; Zeng, Y.; Song, W.; Gaur, U.; Yang, M. 14-3-3 Proteins Are on the Crossroads of Cancer, Aging, and Age-Related Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Y.; Ye, F.; He, X.X. The role of YWHAZ in cancer: A maze of opportunities and challenges. J. Cancer 2020, 11, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J.M.; Murphy, J.; Stomski, F.C.; Berndt, M.C.; Lopez, A.F. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J. Biol. Chem. 2003, 278, 36323–36327. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- MacLaine, N.J.; Hupp, T.R. How phosphorylation controls p53. Cell Cycle (Georget. Tex.) 2011, 10, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Smeenk, L.; van Heeringen, S.J.; Koeppel, M.; Gilbert, B.; Janssen-Megens, E.; Stunnenberg, H.G.; Lohrum, M. Role of p53 serine 46 in p53 target gene regulation. PLoS ONE 2011, 6, e17574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, M.; Flores, E.R. The p53 family orchestrates the regulation of metabolism: Physiological regulation and implications for cancer therapy. Br. J. Cancer 2017, 116, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.R.; Stanford, K.I.; Esko, J.D. Heparan sulfate proteoglycans and triglyceride-rich lipoprotein metabolism. Curr. Opin. Lipidol. 2008, 19, 307–313. [Google Scholar] [CrossRef]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef]
- Li, Y.R.; Yang, W.X. Myosins as fundamental components during tumorigenesis: Diverse and indispensable. Oncotarget 2016, 7, 46785–46812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Yan, J.; Chen, W.; Du, P.; Xie, J.; Yu, H.; Wu, H. Knockdown of Myosin VI Inhibits Proliferation of Hepatocellular Carcinoma Cells In Vitro. Chem. Biol. Drug Des. 2015, 86, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Chen, J.; Zhang, X.; Zhai, Y.; Qing, X.; Xing, W.; Zhang, L.; Malik, Y.S.; Yu, H.; Zhu, X. Elevated expression of myosin X in tumours contributes to breast cancer aggressiveness and metastasis. Br. J. Cancer 2014, 111, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouderkirk-Pecone, J.L.; Goreczny, G.J.; Chase, S.E.; Tatum, A.H.; Turner, C.E.; Krendel, M. Myosin 1e promotes breast cancer malignancy by enhancing tumor cell proliferation and stimulating tumor cell de-differentiation. Oncotarget 2016, 7, 46419–46432. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Deng, J.; Zhou, S.; Tao, T.; Su, Q.; Yang, X.; Yang, X. The role of Clusterin in cancer metastasis. Cancer Manag. Res. 2019, 11, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Thomas, G.; Volarević, S. Corrigendum: Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Turi, Z.; Lacey, M.; Mistrik, M.; Moudry, P. Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 2019, 11, 2512–2540. [Google Scholar] [CrossRef]
- Bursać, S.; Prodan, Y.; Pullen, N.; Bartek, J.; Volarević, S. Dysregulated Ribosome Biogenesis Reveals Therapeutic Liabilities in Cancer. Trends Cancer 2021, 7, 57–76. [Google Scholar] [CrossRef]
- Iadevaia, V.; Liu, R.; Proud, C.G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Hannan, K.M.; Riddell, K.; Ng, P.Y.; Peck, A.; Lee, R.S.; Hung, S.; Astle, M.V.; Bywater, M.; Wall, M.; et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci. Signal. 2011, 4, ra56. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhu, H.; Gu, M.; Luo, Q.; Ding, J.; Yao, Y.; Chen, F.; Wang, Z. DHTKD1 is essential for mitochondrial biogenesis and function maintenance. FEBS Lett. 2013, 587, 3587–3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycan Signaling in Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 6588. [Google Scholar] [CrossRef]
- van Kuppevelt, T.H.; Dennissen, M.A.; van Venrooij, W.J.; Hoet, R.M.; Veerkamp, J.H. Generation and application of type-specific anti-heparan sulfate antibodies using phage display technology. Further evidence for heparan sulfate heterogeneity in the kidney. J. Biol. Chem. 1998, 273, 12960–12966. [Google Scholar] [CrossRef] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Name of Primer | Sequence (5′-3′ Orientation) | Tm (°C) |
|---|---|---|---|
| Reverse | SJ2* | GTGGAGGCAGCTGTA | 50.9 |
| Forward | Albumin promoter | GGCAAACATACGCAAGGGA | 55.8 |
| Mice | Male | Female | Total |
|---|---|---|---|
| hSDC1+/+ transgenic DEN | 22 | 18 | 40 |
| hSDC1+/+ transgenic control | 24 | 19 | 43 |
| C57 Black DEN | 14 | 12 | 26 |
| C57 Black control | 8 | 8 | 16 |
| Gene Name | Protein Name | Fold Change | p-Value |
|---|---|---|---|
| Ehhadh | Peroxisomal bifunctional enzyme | 17.5 | 0.0070 |
| Fasn | Fatty acid synthase | 12.0 | 0.0012 |
| Acly | ATP-citrate synthase | 11.6 | 0.0034 |
| Aldh3a2 | Fatty aldehyde dehydrogenase | 7.6 | 0.0057 |
| Acaca | Acetyl-CoA carboxylase 1 | 5.0 | 0.0437 |
| Aadac | Arylacetamide deacetylase | 4.3 | 0.0002 |
| Aacs | Acetoacetyl-CoA synthetase | 3.9 | 0.0326 |
| Mttp | Microsomal triglyceride transfer protein | 3.9 | 0.0266 |
| Gcdh | Glutaryl-CoA dehydrogenase | 3.5 | 0.0185 |
| Apoa1 | Apolipoprotein A-I | 3.5 | 0.0012 |
| Decr1 | 2,4-dienoyl-CoA reductase | 3.1 | 0.0085 |
| Gene Family | Family Members in WT DEN Group | Family Members in hSDC1+/+ DEN Group |
|---|---|---|
| Ribosome | ||
| Eif | 3c; 5 | 3k; 3l; 4h |
| Tpt | - | 1 |
| Rpl | 19; 38 | 28; 34; 35; 35a; p1 |
| Rps | 15; 20; 23 | 10; 26; 28 |
| Srp | 68; r | - |
| Proteasome | ||
| Psm | d11; d14; e1 | a6; b2; b6; b7; c3; d3; d4 |
| Adrm | 1 | - |
| Numb | - | ✓ |
| Mitochondrial respiratory chain | ||
| Atp | - | 5j |
| Cox | - | 6c |
| Nduf | - | b8; a6; a7; b7; |
| Mtch | - | 2 |
| mt-Nd | - | 5 |
| Myosin | ||
| Myoz | 1 | - |
| Myh | 1; 4; 7; 8 | - |
| Tpm | 2 | - |
| Myl | 1; pf | - |
| Myb | pc2 | - |
| Actin | ||
| Actn | 2; 3 | - |
| Troponin | ||
| Tnn | c2; i2; t3 | - |
| HDL formed cluster | ||
| Apo | a1; a4; b | - |
| Pzp | ✓ | - |
| Serpin | a1b; a1d | - |
| Hrg | ✓ | - |
| Ahsg | ✓ | - |
| Miscellaneous | - | Elavl1; Hnrnpa0-1; Snrnp200; U2af2; Matr3; Anp32a |
| Gene Family | Family Members in WT DEN Group | Family Members in hSDC1+/+ DEN Group |
|---|---|---|
| Ribosome | ||
| Eif | 2a; 3g; 3i; 4g1; 4h | 3b; 3f; 4b |
| Rpl | 18a; 19; p1 | 13a; 27; 26; 28; 31; 34; 35a; 37a |
| Rps | - | 5; 24; 26 |
| Eef | - | 1a2 |
| Proteasome | ||
| Psm | c3; d4; d11; d14 | b5; b6; d12 |
| Numb | - | ✓ |
| Mitochondrial respiratory chain | ||
| Atp | - | 5d; 5c1; 6v1a |
| Cox | - | 7c |
| Nduf | - | b10; s4 |
| Mtch | - | 2 |
| mt-Nd | - | 5 |
| Myosin-actin | ||
| Myh | - | 1; 3; 4; 7; 8; 11; 14; |
| Myl | - | 1; 3; pf |
| Actn | - | 2; 3 |
| Tnn | - | c3 |
| Tpm | - | 2 |
| Des | - | ✓ |
| Atp | - | 2a1 |
| Fat net | ||
| Apo | a1; a4; c2; e; h | - |
| Pzp | ✓ | - |
| Serpin | a1b; a1c; c1; f2 | - |
| Calu | ✓ | - |
| Ahsg | ✓ | - |
| Anx | a1; a2 | - |
| Ola | 1 | - |
| Plg | ✓ | - |
| Mttp | ✓ | - |
| Clu | ✓ | - |
| Kng | 1 | - |
| mRNA splicing | ||
| Srsf | 1; 7 | - |
| Prp | 18 | - |
| Endoplasmic retic stress—protein folding | ||
| Dnaj | a2; b11; c3 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reszegi, A.; Karászi, K.; Tóth, G.; Rada, K.; Váncza, L.; Turiák, L.; Schaff, Z.; Kiss, A.; Szilák, L.; Szabó, G.; et al. Overexpression of Human Syndecan-1 Protects against the Diethylnitrosamine-Induced Hepatocarcinogenesis in Mice. Cancers 2021, 13, 1548. https://doi.org/10.3390/cancers13071548
Reszegi A, Karászi K, Tóth G, Rada K, Váncza L, Turiák L, Schaff Z, Kiss A, Szilák L, Szabó G, et al. Overexpression of Human Syndecan-1 Protects against the Diethylnitrosamine-Induced Hepatocarcinogenesis in Mice. Cancers. 2021; 13(7):1548. https://doi.org/10.3390/cancers13071548
Chicago/Turabian StyleReszegi, Andrea, Katalin Karászi, Gábor Tóth, Kristóf Rada, Lóránd Váncza, Lilla Turiák, Zsuzsa Schaff, András Kiss, László Szilák, Gábor Szabó, and et al. 2021. "Overexpression of Human Syndecan-1 Protects against the Diethylnitrosamine-Induced Hepatocarcinogenesis in Mice" Cancers 13, no. 7: 1548. https://doi.org/10.3390/cancers13071548