
Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
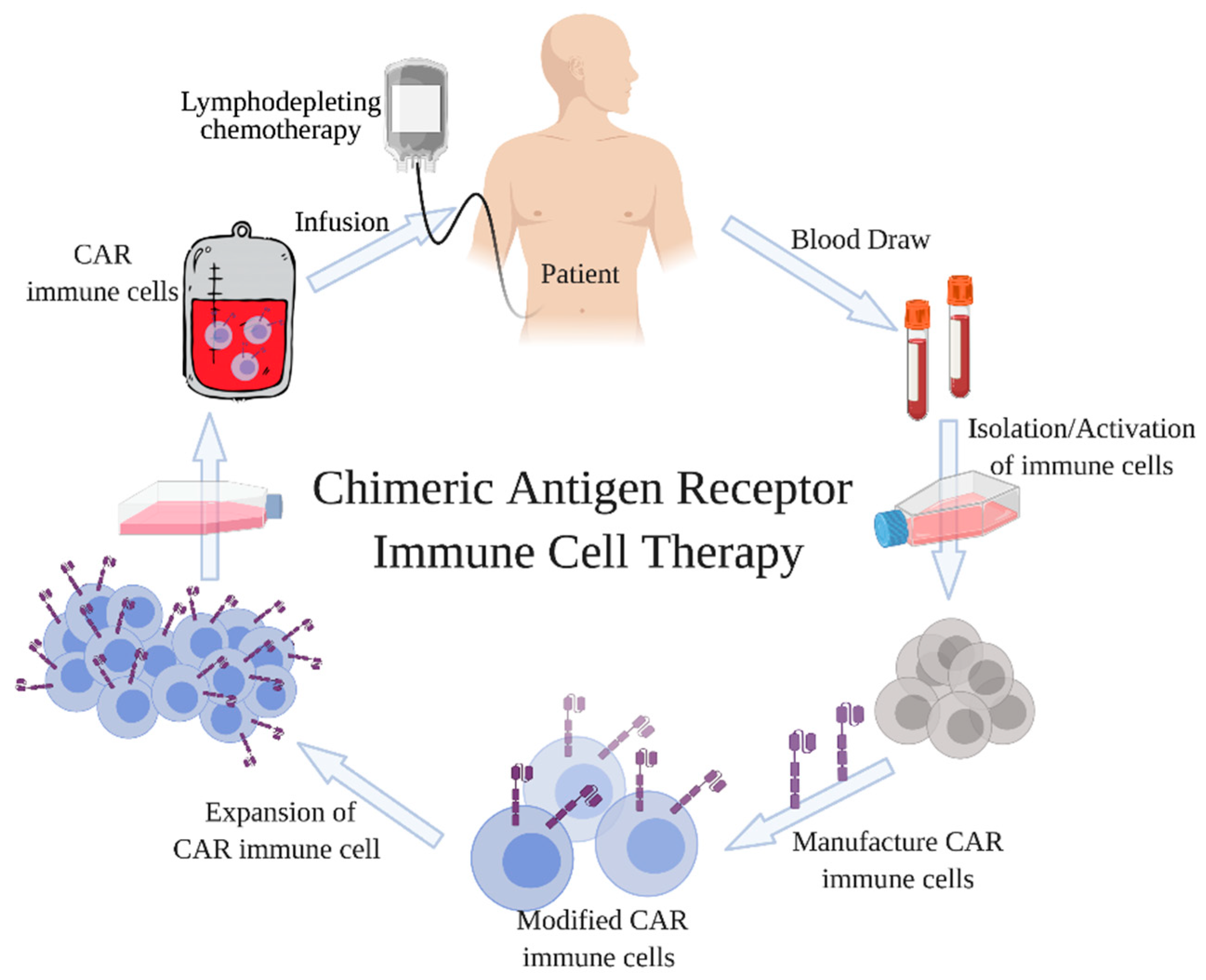
1. Introduction
2. Challenges of CAR T Cell Therapy
3. CAR T Cell Immunotherapy for Solid Tumors
3.1. Impact of TME on T Cell Metabolism
3.2. Metabolic Engineering of CAR Cells
4. Other Potent CAR Immune Cells
4.1. NK Cell CAR Therapies
4.2. CAR Macrophages
5. Summary
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations:
| ACT | Adoptive cell transfer |
| AKT | protein kinase B |
| APCs | Antigen presenting cells |
| ATP | Adenosine triphosphate |
| B-ALL | B cell acute lymphoblastic leukemia |
| Bcl-X(L) | B-cell lymphoma-extra large |
| CAFs | Cancer-associated fibroblasts |
| CAR | Chimeric antigen receptor |
| CAR-M | Chimeric antigen receptor macrophage |
| CAR-Ps | Chimeric antigen receptors for phagocytosis |
| CAT | Catalase |
| CCR | Chimeric costimulatory receptor |
| CLL | Chronic lymphocytic leukemia |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CRS | Cytokine release syndrome |
| ECM | Extracellular matrix |
| ETBR | Endothelin B receptor |
| FAO | Fatty acid oxidation |
| GLUT1 | Glucose transporter 1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HK | Hexokinase 2 |
| HPSE | Heparinase |
| HSPG | Heparan sulphate proteoglycans |
| ICOS | Inducible T cell co-stimulator |
| IDO | Indoleamine-2,3-dioxygenase |
| IL-10 | Interleukin 10 |
| LBCL | Large B cell lymphoma |
| MDSC | Myeloid-derived suppressor cells |
| MICA | Major histocompatibility complex class I polypeptide-related sequence A |
| NK | Natural killer |
| OXPHOS | Oxidative phosphorylation |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PFK-6 | phosphofructo-2-kinase |
| PGC1-α | PPAR-gamma coactivator 1-α |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide 3-kinase |
| PSMA | Prostate-specific membrane |
| ROS | Reactive oxygen species |
| ScFv | Single chain variable fragment |
| TAA | Tumor-associated antigens |
| TCR | T cell receptor |
| TGFβ | Transforming growth factor beta |
| TILs | Tumor-infiltration lymphocytes |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
References
- Batlevi, C.L.; Matsuki, E.; Brentjens, R.J.; Younes, A. Novel immunotherapies in lymphoid malignancies. Nat. Rev. Clin. Oncol. 2016, 13, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/results/details?cond=CAR+T+cells (accessed on 29 June 2020).
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat. Rev. Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. J. Immunol. 2009, 184, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Prendergast, G.C. Marrying immunotherapy with chemotherapy: Why say IDO? Cancer Res. 2005. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Huang, R.R.; Wen, D.R.; Paul, E.; Wünsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R. Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies Against Cancer. Front. Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Heitzeneder, S.; Mackall, C.L. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell. 2017, 31, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from car t-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M. CAR therapy: The CD19 paradigm. J. Clin. Investig. 2015, 125, 3392–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; Storm, J.; Barrett, D.M. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Heinmöller, P.; Gross, C.; Beyser, K.; Schmidtgen, C.; Maass, G.; Pedrocchi, M.; Rüschoff, J. HER2 Status in Non-Small Cell Lung Cancer: Results from Patient Screening for Enrollment to a Phase II Study of Herceptin. Clin Cancer Res. 2003, 9, 5238–5243. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 1–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Xu, H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.V.; Porter, D.L. CAR T-cells merge into the fast lane of cancer care. Am. J. Hematol. 2016, 91, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, B.; Moyes, J.S.; Huls, H.; Cooper, L.J.N. Driving CAR-Based T-Cell Therapy to Success. Curr. Hematol. Malig Rep. 2014, 9, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.E.; Debets, R.; Pule, M.; Hawkins, R.E.; Abken, H. CAR–T cells and solid tumors: Tuning T cells to challenge an inveterate foe. Trends Mol. Med. 2012, 18, 377–384. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors review-article. Cell Death Dis. 2018. [Google Scholar] [CrossRef]
- Irving, M.; de Silly, R.V.; Scholten, K.; Dilek, N.; Coukos, G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: Don’t forget the fuel. Front. Immunol. 2017, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Hu, Q.; Dukhovlinova, E.; Chen, G.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv. Mater. 2019, 31, 1900192. [Google Scholar] [CrossRef]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Rataj, F.; Jacobi, S.J.; Stoiber, S.; Asang, F.; Ogonek, J.; Tokarew, N.; Cadilha, B.L.; Puijenbroek, E.V.; Heise, C.; Duewell, P.; et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 2019, 120, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Clémenceau, B.; Congy-Jolivet, N.; Gallot, G.; Vivien, R.; Gaschet, J.; Thibault, G.; Vié, H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006, 107, 4669–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Ochi, F.; Fujiwara, H.; Tanimoto, K.; Asai, H.; Miyazaki, Y.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Barrett, J.; et al. Gene-modified human α/β-T cells expressing a chimeric CD16-CD3ζ receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol. Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Fuertes-Marraco, S.A.; Legat, A.; Meyer, C.; Speiser, D.E. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012, 33, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Kouidhi, S.; Elgaaied, A.B.; Chouaib, S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front. Immunol. 2017, 8, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017, 32, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef]
- Herbel, C.; Patsoukis, N.; Bardhan, K.; Seth, P.; Weaver, J.D.; Boussiotis, V.A. Clinical significance of T cell metabolic reprogramming in cancer. Clin. Transl. Med. 2016, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.C.; Liu, P.S. Metabolic communication in tumors: A new layer of immunoregulation for immune evasion. J. Immunother. Cancer 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Weaver, J.D.; Strauss, L.; Herbel, C.; Seth, P.; Boussiotis, V.A. Immunometabolic regulations mediated by coinhibitory receptors and their impact on T cell immune responses. Front. Immunol. 2017, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; MacIntyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; Van Der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, H.C.; Salmond, R.J. Targeting the tumor microenvironment and T cell metabolism for effective cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate regulates metabolic and proinflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Lind, D.S. Arginine and cancer. J. Nutr. 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.P.; Sinha, M.; Ratay, M.L.; Ding, X.; Balmert, S.C.; Workman, C.J.; Wang, Y.; Vignali, D.A.A.; Little, S.R. Localized Multi-Component Delivery Platform Generates Local and Systemic Anti-Tumor Immunity. Adv. Funct. Mater. 2017, 27. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Kolev, M.; Dimeloe, S.; Le Friec, G.; Navarini, A.; Arbore, G.; Povoleri, G.A.; Fischer, M.; Belle, R.; Loeliger, J.; Develioglu, L.; et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015, 42, 1033–1047. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity Cell Press 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; Pearce, E.L. Targeting T cell metabolism for therapy. Trends Immunol. 2015, 36, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; DeSantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulè, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; Mcgettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, J.; Keep, O.; Senovilla, L.; Lissa, D.; Castedo, M.; Kroemer, G.; Galluzzi, L. Functions of BCL-XL at the interface between cell death and metabolism. Int. J. Cell Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Abate-Daga, D.; Davila, M.L. CAR models: Next-generation CAR modifications for enhanced T-cell function. Mol. Ther. Oncolytics 2016, 3, 16014. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Kumar, A.; Chamoto, K. Immune metabolism in PD-1 blockade-based cancer immunotherapy. Int. Immunol. 2021, 33, 17–26. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Xu, B.; Zhu, N.; Ren, B. PGC-1α activator–induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori 2020. [Google Scholar] [CrossRef]
- Yee, C. Adoptive T cell therapy: Points to consider. Curr. Opin. Immunol. 2018, 51, 197–203. [Google Scholar] [CrossRef]
- Le Bourgeois, T.; Strauss, L.; Aksoylar, H.I.; Daneshmandi, S.; Seth, P.; Patsoukis, N.; Boussiotis, V.A. Targeting T cell metabolism for improvement of cancer immunotherapy. Front. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.P.; Dolgova, N.V.; Clare-Salzler, M.J.; Keselowsky, B.G. Adhesive substrate-modulation of adaptive immune responses. Biomaterials 2008, 29, 4736–4750. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.P.; Carstens, M.R.; Lewis, J.S.; Dolgova, N.; Xia, C.Q.; Clare-Salzler, M.J.; Keselowsky, B.G. A cell-based microarray to investigate combinatorial effects of microparticle-encapsulated adjuvants on dendritic cell activation. J. Mater. Chem. 2016, 4, 1672–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, A.P.; Clare-Salzler, M.J.; Keselowsky, B.G. A high-throughput microparticle microarray platform for dendritic cell-targeting vaccines. Biomaterials 2009, 30, 4168–4177. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- A Morrissey, M.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Mangal, J.L.; Inamdar, S.; Yang, Y.; Dutta, S.; Wankhede, M.; Shi, X.; Gu, H.; Green, M.D.; Rege, K.; Curtis, M.; et al. Metabolite releasing polymers control dendritic cell function by modulating their energy metabolism. J. Mater. Chem. B 2020, 8, 5195–5203. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.A.; Oldridge, D.; Wu, G.; Sussman, R.T.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, A.; Cui, H.; Caligiuri, M.A.; Yu, J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 1–22. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Gobius, I.; Souza-Fonseca-Guimaraes, F. Natural killer cell engineering—A new hope for cancer immunotherapy. Semin. Hematol. 2020, 57, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Del Principe, M.I.; Lauro, D.; et al. In vitro elimination of epidermal growth factor receptor-overexpressing cancer cells by CD32A-chimeric receptor T cells in combination with cetuximab or panitumumab. Int. J. Cancer 2020, 146, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, L.O.; Adeshakin, A.O.; Sani, M.M.; Bi, J.; Wan, X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology 2019, 158, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, srep39833. [Google Scholar] [CrossRef]
- Navin, I.; Lam, M.T.; Parihar, R. Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers 2020, 12, 3871. [Google Scholar] [CrossRef]
- Rautela, J.; Surgenor, E.; Huntington, N.D. Efficient genome editing of human natural killer cells by CRISPR RNP. bioRxiv 2018. [Google Scholar] [CrossRef]
- Sekiba, K.; Yamagami, M.; Otsuka, M.; Suzuki, T.; Kishikawa, T.; Ishibashi, R.; Ohno, M.; Sato, M.; Koike, K. Transcriptional activation of the MICA gene with an engineered CRISPR-Cas9 system. Biochem. Biophys. Res. Commun. 2017, 486, 521–525. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.H.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modiWed eVectors that carry a tumor-speciWc antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Hu, Y.; Tian, Z.-G.; Zhang, C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol. Sin. 2017, 39, 167–176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| TME Factors Affecting Immune Cell Metabolism | Direct and Indirect Metabolic Impairment of Immune Cells in TME | Strategy to Overcome Induced Metabolic Impairment | References |
|---|---|---|---|
| Increased hypoxia | Increases HIF1-a and PD-L1 on MDSCs for T cell exhaustion and Treg generation | Anti-PD-L1 and HIF inhibitors | [43,69,70] |
| Increased reactive oxygen species | Oxidative stress-mediated inhibition of NF-kB or mTOR for the prevention of T cell activation | Catalase, an antioxidant enzyme | [32,42] |
| Decreased glucose availability | Reduced AKT, mTOR, GLUT1, phosphoenolpyruvate, and increased PD-1 expression in T cells | anti-PD-1, anti-PD-L1, anti-CTLA-4 | [27,46,48,49,50] |
| Increased lactate | Inhibition of T cell glycolysis and function | Blocking acidification prior to anti-PD-1 or anti-CTLA-4 administration | [52] |
| Low levels of arginine | Reduced responsiveness of T cells due to decreased expression of CD3ζ chain | Administering arginine | [55,56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangal, J.L.; Handlos, J.L.; Esrafili, A.; Inamdar, S.; Mcmillian, S.; Wankhede, M.; Gottardi, R.; Acharya, A.P. Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies. Cancers 2021, 13, 1123. https://doi.org/10.3390/cancers13051123
Mangal JL, Handlos JL, Esrafili A, Inamdar S, Mcmillian S, Wankhede M, Gottardi R, Acharya AP. Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies. Cancers. 2021; 13(5):1123. https://doi.org/10.3390/cancers13051123
Chicago/Turabian StyleMangal, Joslyn L., Jamie L. Handlos, Arezoo Esrafili, Sahil Inamdar, Sidnee Mcmillian, Mamta Wankhede, Riccardo Gottardi, and Abhinav P. Acharya. 2021. "Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies" Cancers 13, no. 5: 1123. https://doi.org/10.3390/cancers13051123