
FAM72, Glioblastoma Multiforme (GBM) and Beyond

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
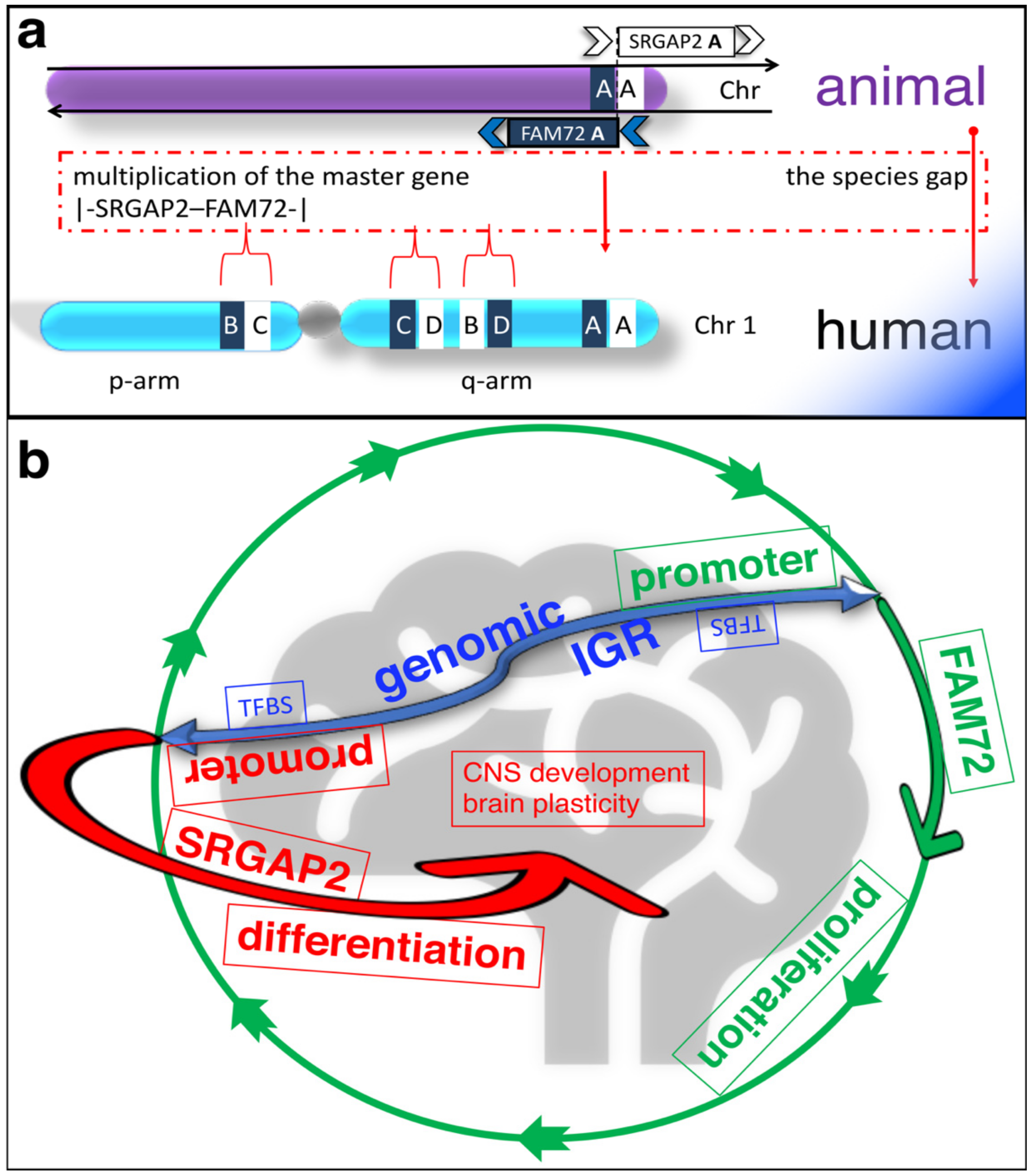
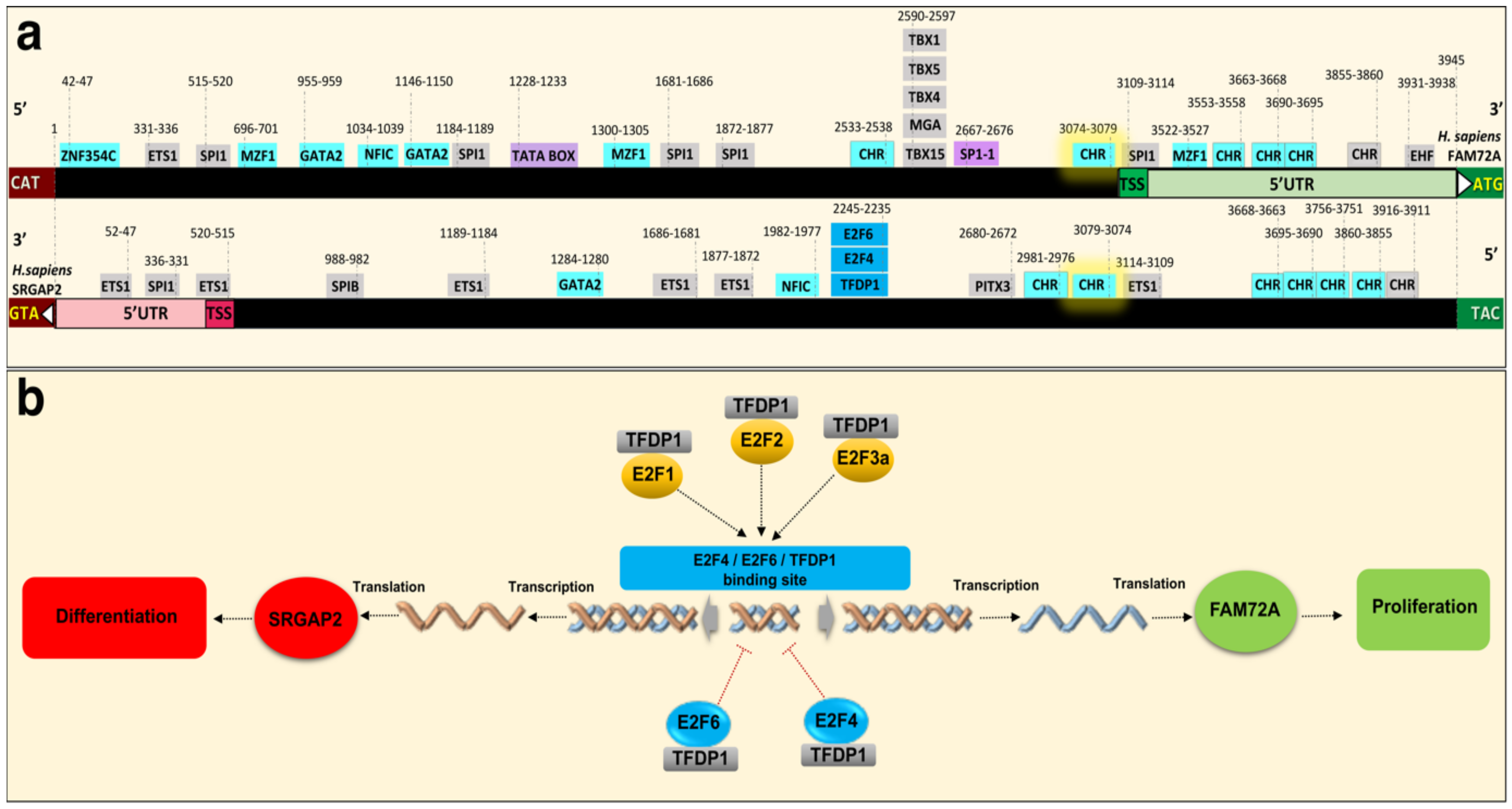
2. Physiological Function of the |-SRGAP2–FAM72-| Master Gene
3. Pathophysiological Function of the |-SRGAP2–FAM72-| Master Gene—FAM72 Expression in Various Types of Cancer
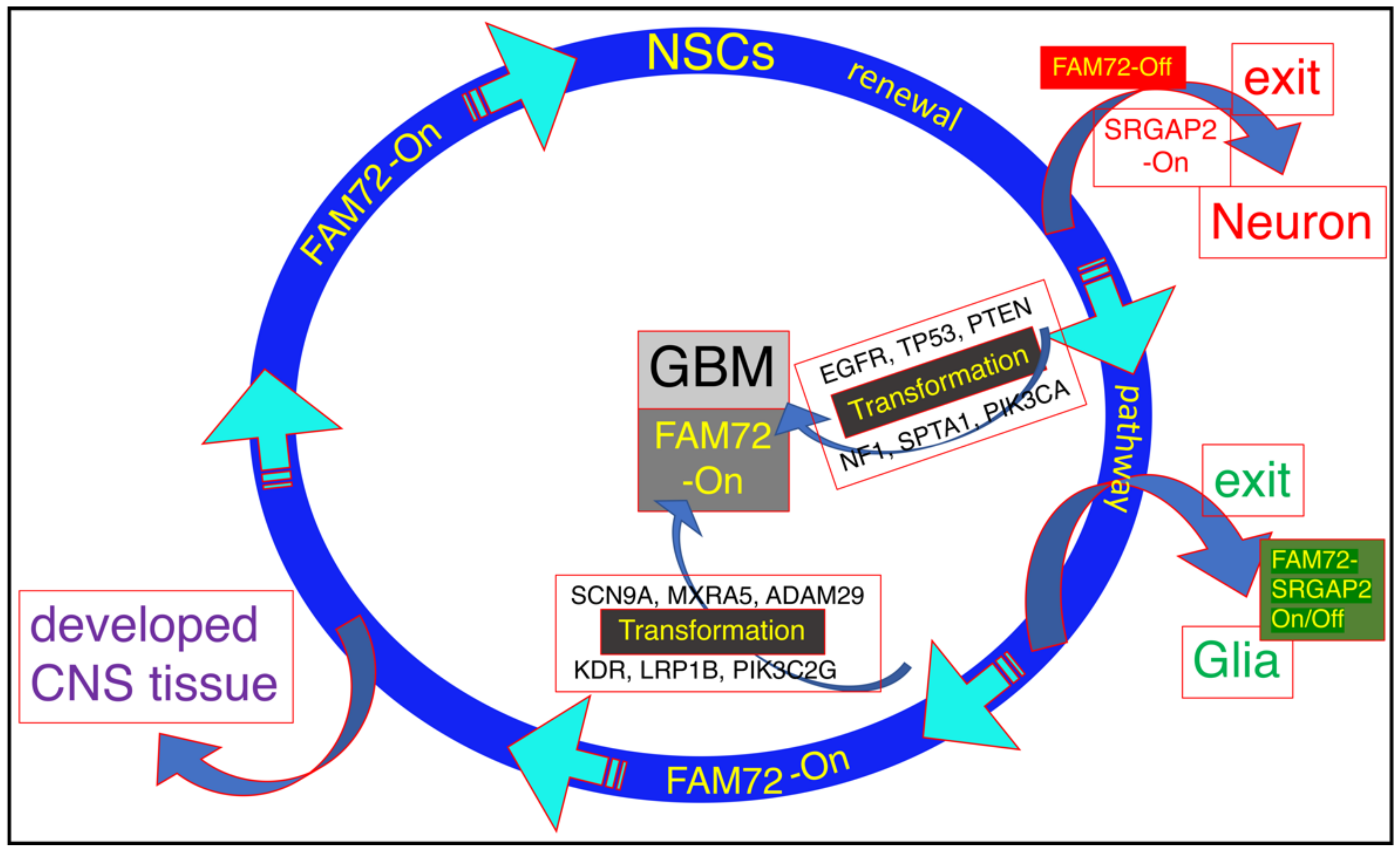
3.1. The |-SRGAP2–FAM72-| Master Gene in Brain Cancer
3.2. The |-SRGAP2–FAM72-| Master Gene in Other Cancerous Tissues
3.3. FAM72 in Adrenocortical Carcinoma
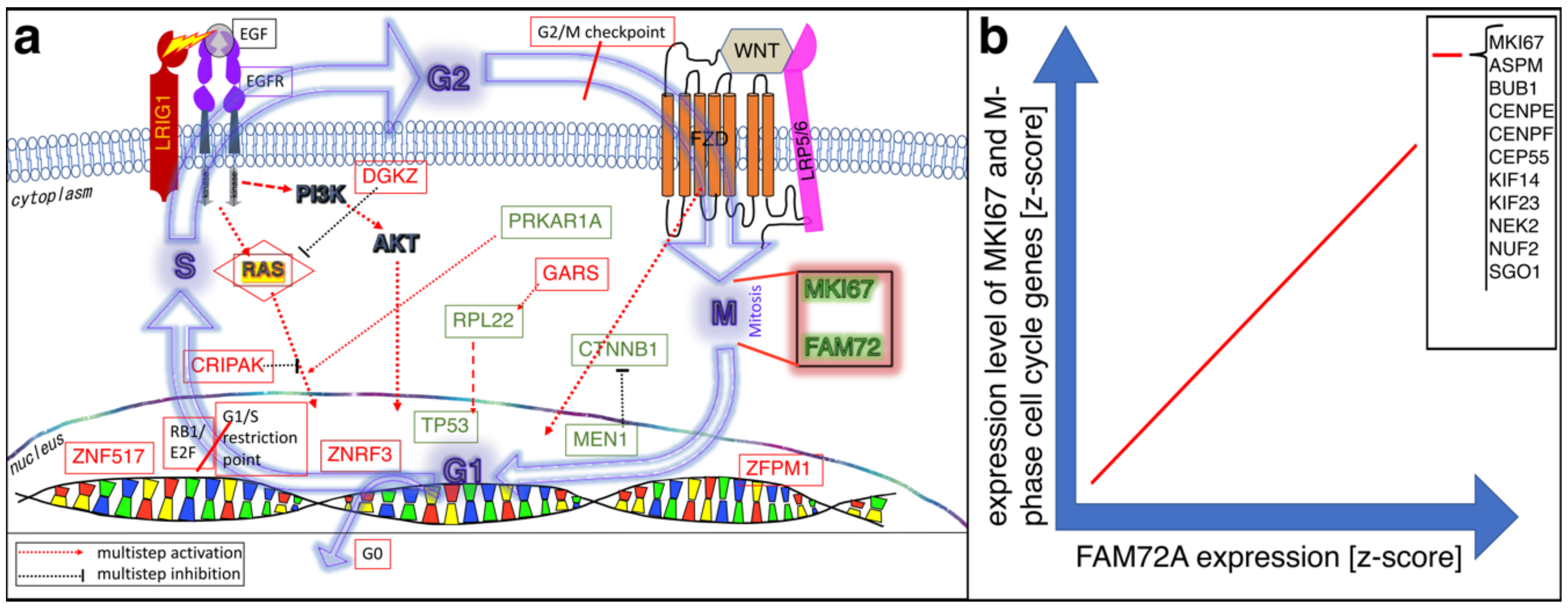
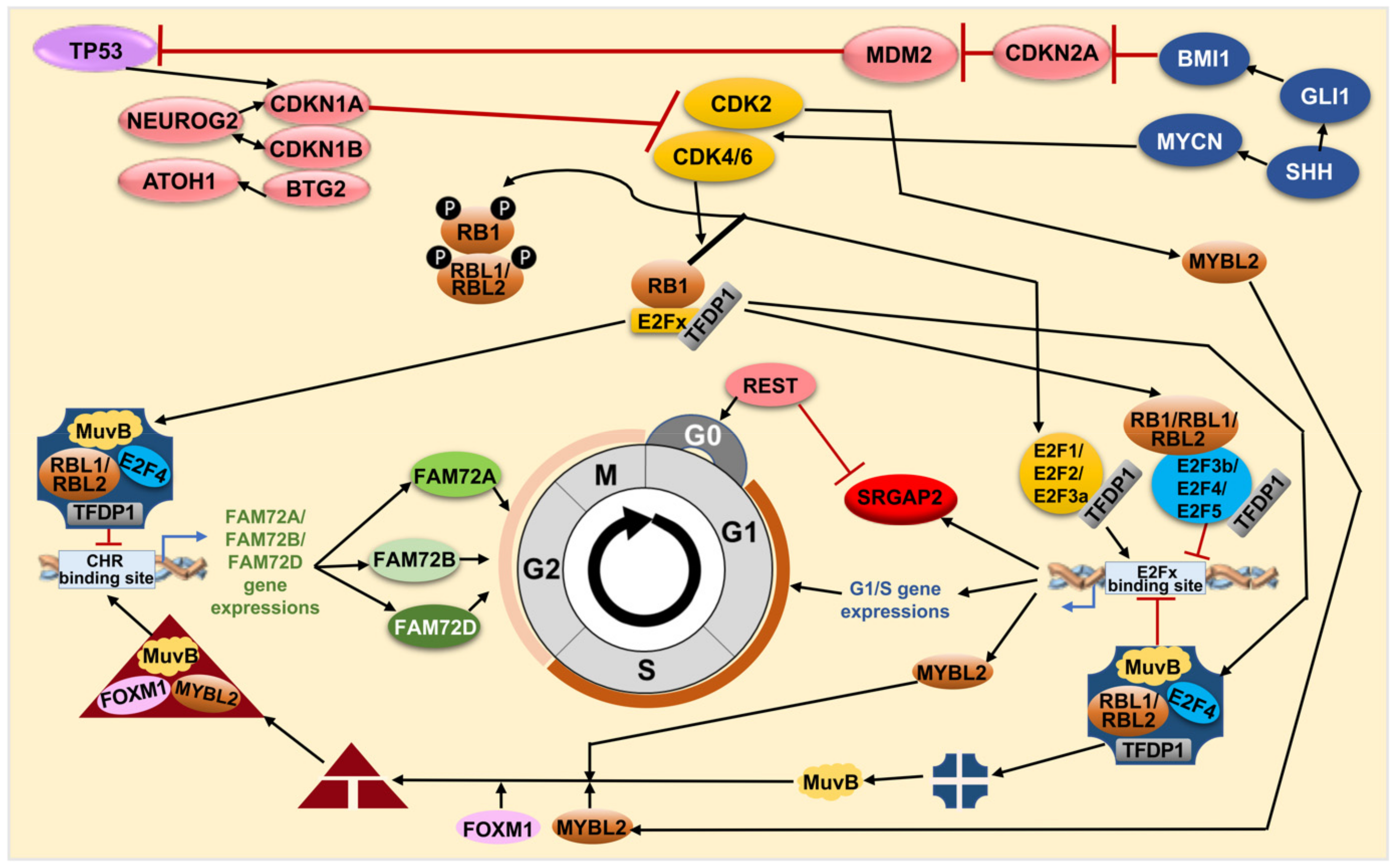
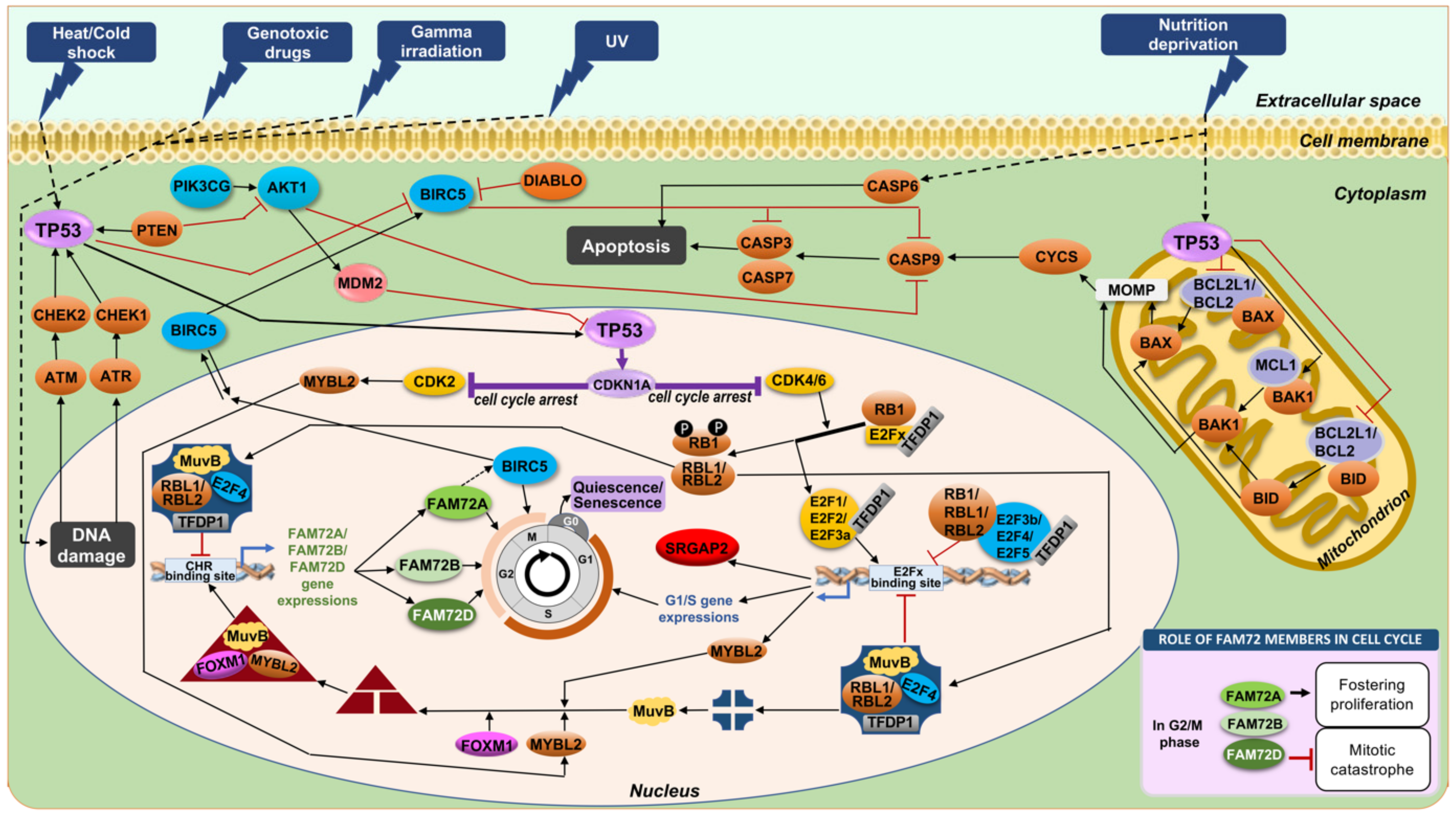
4. FAM72 and Its Role in the Cell Cycle
4.1. FAM72 in the M-Phase of the Cell Cycle
4.2. FAM72 in the G0 Stage of the Cell Cycle
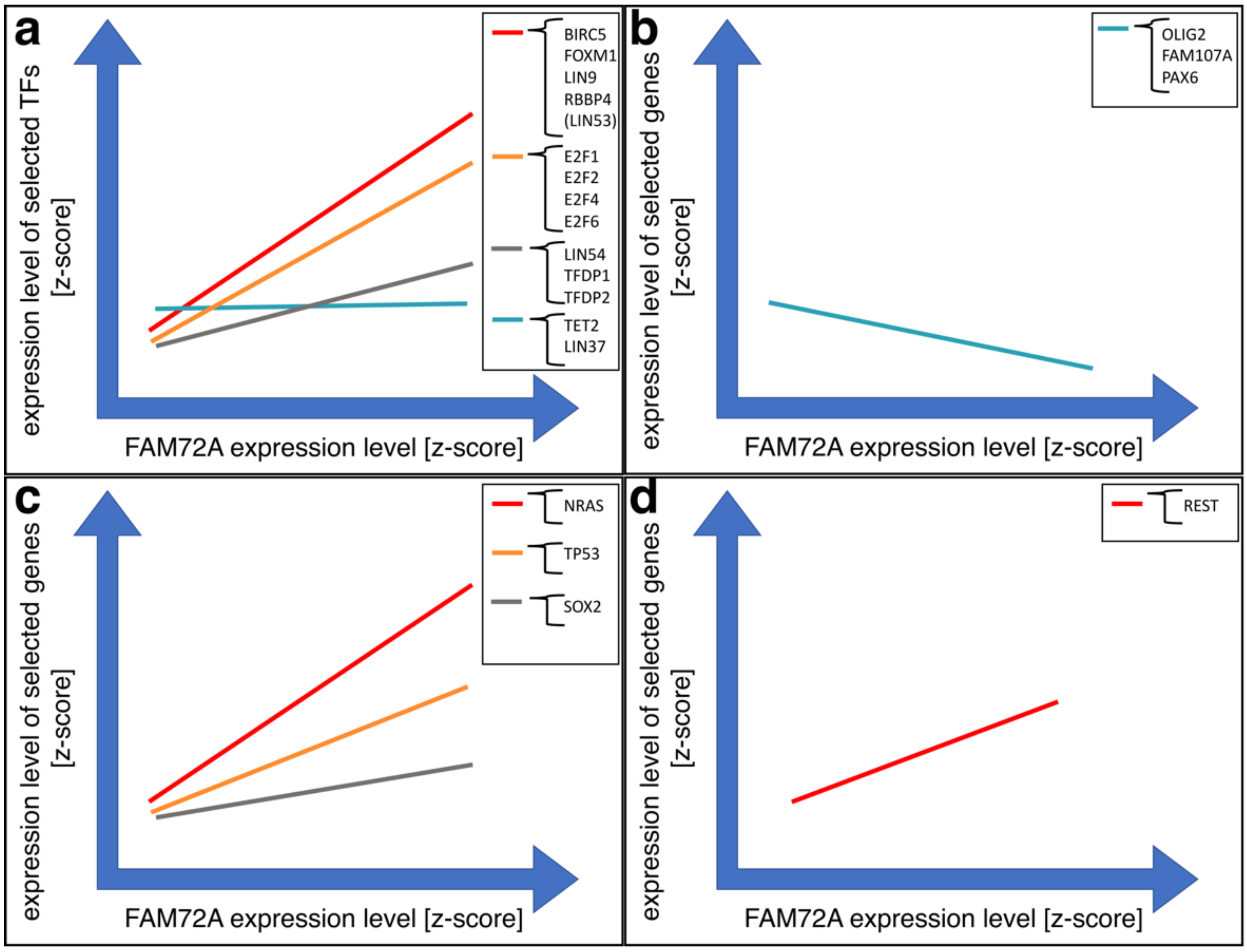
4.3. Governance of FAM72 Expression: The IGR and Its TFBSs
4.4. FAM72 Expression and the RE1 Silencing Transcription Factor
4.5. FAM72 Expression and Long Non-Coding RNAs
4.6. Anti-Apoptotic Features of |-SRGAP2–FAM72-| via TP53
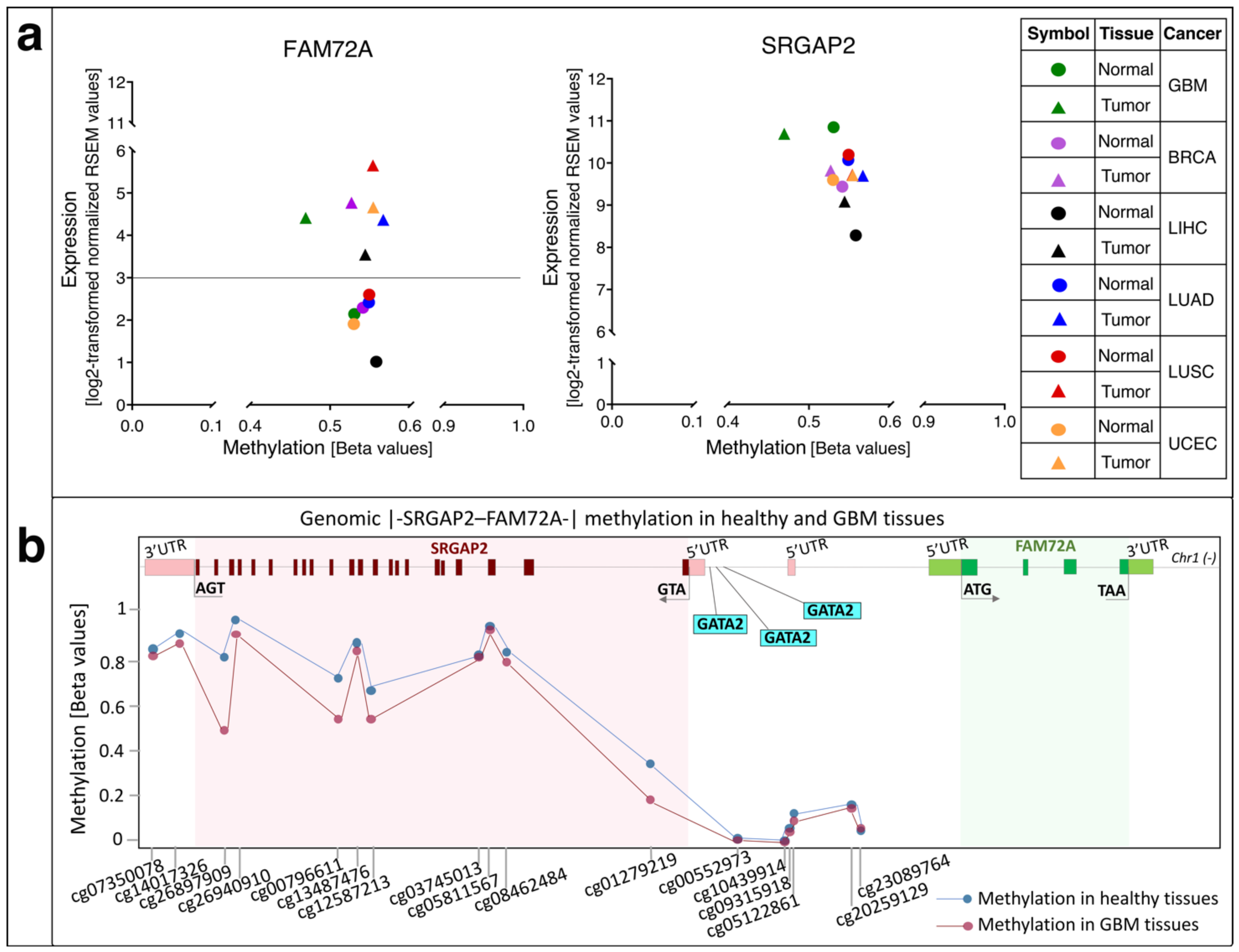
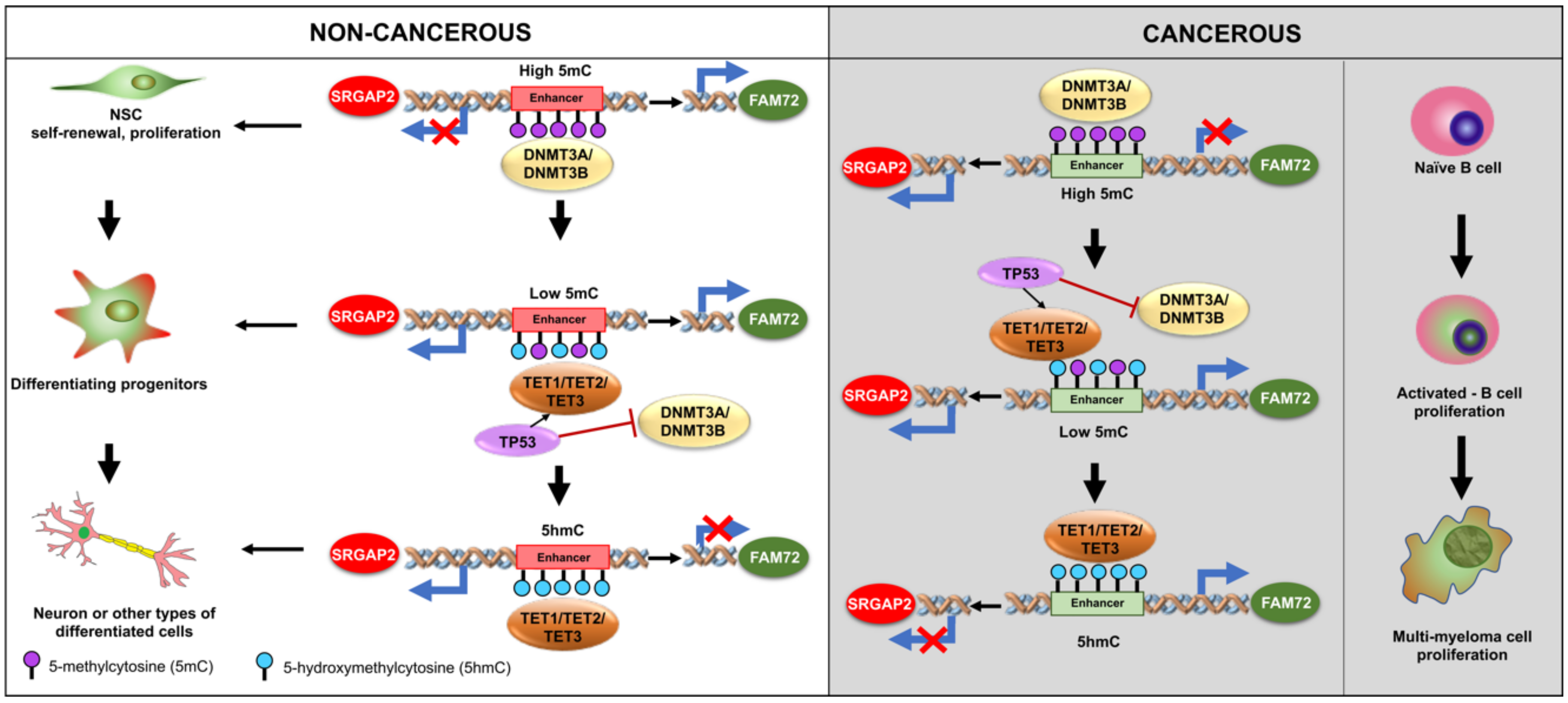
5. Methylation of FAM72 in Cancerous Tissues


6. FAM72 and FAM107A in GBM
7. FAM72 and Its Role as a Potential Biomarker in Clinical Cancer Diagnostics
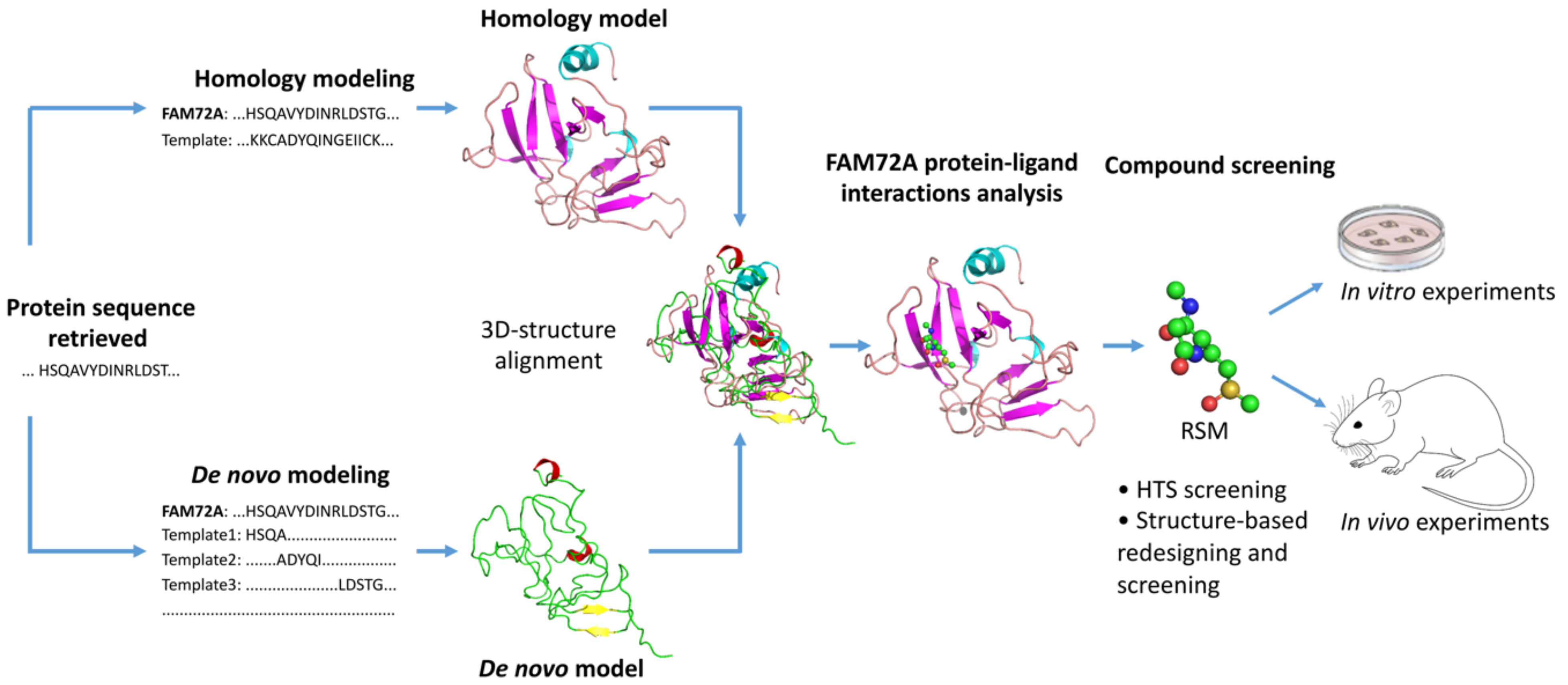
8. FAM72 and Its Role in Cancer Therapy: Therapeutic Options against Tumorigenic FAM72
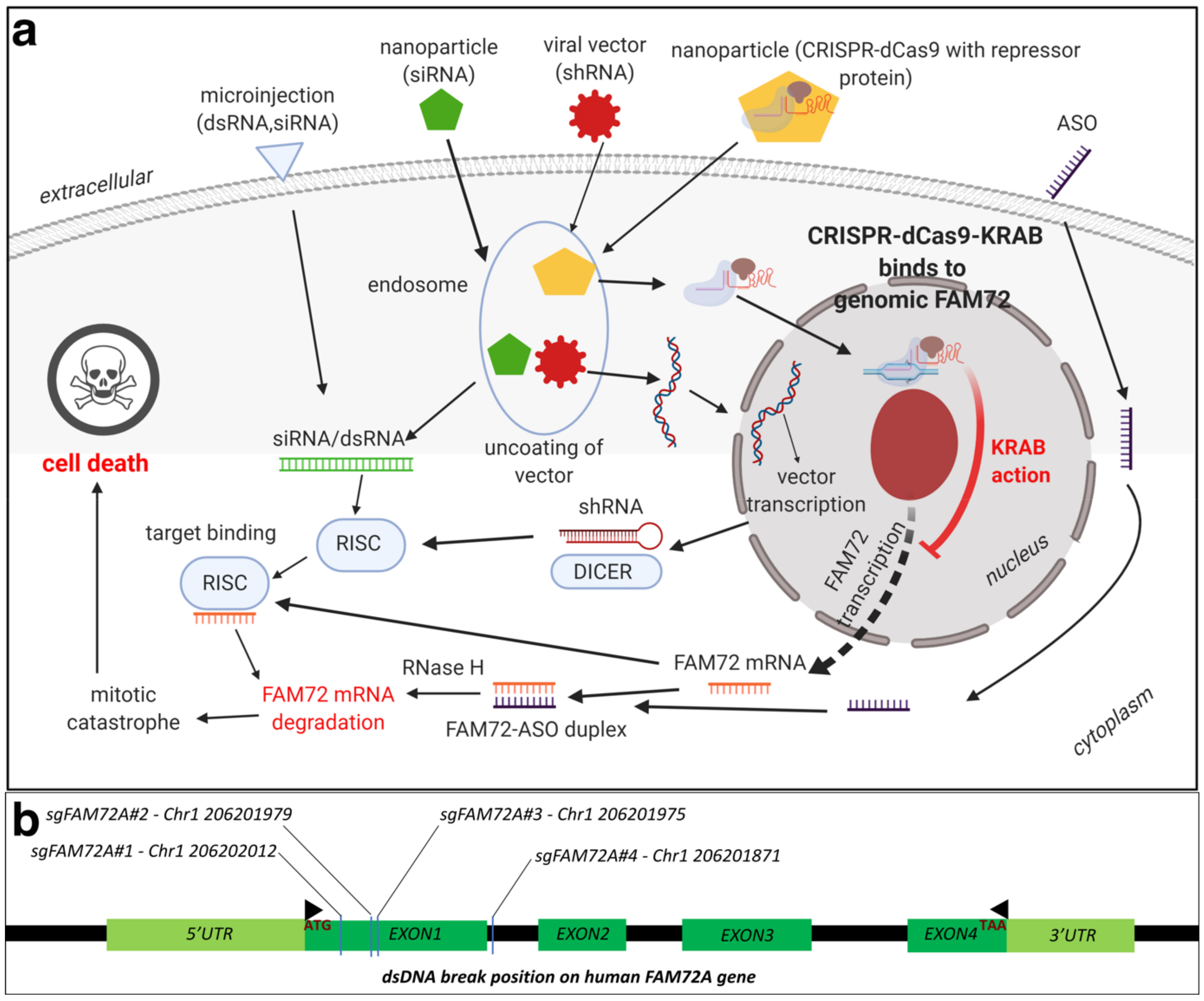
8.1. Therapeutic Options against Tumorigenic FAM72: RNA Interference (RNAi)
8.2. Therapeutic Options against Tumorigenic FAM72: CRISPR-Cas9
8.3. Therapeutic Options against Tumorigenic FAM72: Chemotherapy
9. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Kutzner, A.; Pramanik, S.; Kim, P.S.; Heese, K. All-or-(N)One—An epistemological characterization of the human tumorigenic neuronal paralogous FAM72 gene loci. Genomics 2015, 106, 278–285. [Google Scholar] [CrossRef]
- Ho, N.T.; Kim, P.S.; Kutzner, A.; Heese, K. Cognitive Functions: Human vs. Animal—4:1 Advantage |-FAM72-SRGAP2-|. J. Mol. Neurosci. 2017, 61, 603–606. [Google Scholar] [CrossRef]
- Nehar, S.; Mishra, M.; Heese, K. Identification and characterisation of the novel amyloid-beta peptide-induced protein p17. FEBS Lett. 2009, 583, 3247–3253. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.B.; Mancini, E.; et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014, 158, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.T.T.; Kutzner, A.; Heese, K. Brain plasticity, cognitive functions and neural stem cells: A pivotal role for the brain-specific neural master gene |-SRGAP2-FAM72-|. Biol. Chem. 2017, 399, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.Y.; Nuttle, X.; Sudmant, P.H.; Antonacci, F.; Graves, T.A.; Nefedov, M.; Rosenfeld, J.A.; Sajjadian, S.; Malig, M.; Kotkiewicz, H.; et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 2012, 149, 912–922. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, D.H.; Konopka, G. Neuroscience: Genes and human brain evolution. Nature 2012, 486, 481–482. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.T.T.; Kutzner, A.; Heese, K. A Novel Divergent Gene Transcription Paradigm-the Decisive, Brain-Specific, Neural |-Srgap2-Fam72a-| Master Gene Paradigm. Mol. Neurobiol. 2019, 56, 5891–5899. [Google Scholar] [CrossRef]
- Chen, Y.; Pai, A.A.; Herudek, J.; Lubas, M.; Meola, N.; Jarvelin, A.I.; Andersson, R.; Pelechano, V.; Steinmetz, L.M.; Jensen, T.H.; et al. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat. Genet. 2016, 48, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Lacadie, S.A.; Ibrahim, M.M.; Gokhale, S.A.; Ohler, U. Divergent transcription and epigenetic directionality of human promoters. FEBS J. 2016, 283, 4214–4222. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.M.; Karabacak, A.; Glahs, A.; Kolundzic, E.; Hirsekorn, A.; Carda, A.; Tursun, B.; Zinzen, R.P.; Lacadie, S.A.; Ohler, U. Determinants of promoter and enhancer transcription directionality in metazoans. Nat. Commun. 2018, 9, 4472. [Google Scholar] [CrossRef]
- Rahane, C.S.; Kutzner, A.; Heese, K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J. Neurooncol. 2019, 141, 57–70. [Google Scholar] [CrossRef]
- Schneider, J.; Karpf, J.; Beckervordersandforth, R. Role of Astrocytes in the Neurogenic Niches. In Methods in Molecular Biology; Springer: Berlin, Germany, 2019; Volume 1938, pp. 19–33. [Google Scholar] [CrossRef]
- Cassé, F.; Richetin, K.; Toni, N. Astrocytes’ Contribution to Adult Neurogenesis in Physiology and Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 432. [Google Scholar] [CrossRef] [Green Version]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Charrier, C.; Joshi, K.; Coutinho-Budd, J.; Kim, J.E.; Lambert, N.; de Marchena, J.; Jin, W.L.; Vanderhaeghen, P.; Ghosh, A.; Sassa, T.; et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 2012, 149, 923–935. [Google Scholar] [CrossRef] [Green Version]
- Fossati, M.; Pizzarelli, R.; Schmidt, E.R.; Kupferman, J.V.; Stroebel, D.; Polleux, F.; Charrier, C. SRGAP2 and Its Human-Specific Paralog Co-Regulate the Development of Excitatory and Inhibitory Synapses. Neuron 2016, 91, 356–369. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, D.H.; Rakic, P. Cortical evolution: Judge the brain by its cover. Neuron 2013, 80, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Rincic, M.; Rados, M.; Krsnik, Z.; Gotovac, K.; Borovecki, F.; Liehr, T.; Brecevic, L. Complex intrachromosomal rearrangement in 1q leading to 1q32.2 microdeletion: A potential role of SRGAP2 in the gyrification of cerebral cortex. Mol. Cytogenet. 2016, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, J.; Nedivi, E. Filling the (SR)GAP in Excitatory/Inhibitory Balance. Neuron 2016, 91, 205–207. [Google Scholar] [CrossRef]
- Jiao, Q.; Wang, L.; Zhang, Z.; Wang, Y.; Yan, H.; Ma, W.; Jin, W.; Lu, H.; Liu, Y. Dynamic expression of srGAP2 in cell nuclei and cytoplasm during the differentiation of rat neural stem cells in vitro. Mol. Med. Rep. 2016, 14, 4599–4605. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Zhang, X.; Fink, S.P.; Platzer, P.; Wilson, K.; Willson, J.K.; Wang, Z.; Markowitz, S.D. Ugene, a newly identified protein that is commonly overexpressed in cancer and binds uracil DNA glycosylase. Cancer Res. 2008, 68, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Rajan, P.; Stockley, J.; Sudbery, I.M.; Fleming, J.T.; Hedley, A.; Kalna, G.; Sims, D.; Ponting, C.P.; Heger, A.; Robson, C.N.; et al. Identification of a candidate prognostic gene signature by transcriptome analysis of matched pre- and post-treatment prostatic biopsies from patients with advanced prostate cancer. BMC Cancer 2014, 14, 977. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Fang, C.; Li, X.; Geng, Y.; Li, R.; Wu, C.; Jiang, J.; Wu, C. Predictive analysis of long non-coding RNA expression profiles in diffuse large B-cell lymphoma. Oncotarget 2017, 8, 23228–23236. [Google Scholar] [CrossRef] [Green Version]
- Chatonnet, F.; Pignarre, A.; Serandour, A.A.; Caron, G.; Avner, S.; Robert, N.; Kassambara, A.; Laurent, A.; Bizot, M.; Agirre, X.; et al. The hydroxymethylome of multiple myeloma identifies FAM72D as a 1q21 marker linked to proliferation. Haematologica 2020, 105, 774–783. [Google Scholar] [CrossRef] [Green Version]
- Marko, T.A.; Shamsan, G.A.; Edwards, E.N.; Hazelton, P.E.; Rathe, S.K.; Cornax, I.; Overn, P.R.; Varshney, J.; Diessner, B.J.; Moriarity, B.S.; et al. Slit-Robo GTPase-Activating Protein 2 as a metastasis suppressor in osteosarcoma. Sci. Rep. 2016, 6, 39059. [Google Scholar] [CrossRef]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Rahane, C.S.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet. 2019, 230, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Lippert, J.; Appenzeller, S.; Liang, R.; Sbiera, S.; Kircher, S.; Altieri, B.; Nanda, I.; Weigand, I.; Gehrig, A.; Steinhauer, S.; et al. Targeted Molecular Analysis in Adrenocortical Carcinomas: A Strategy Toward Improved Personalized Prognostication. J. Clin. Endocrinol. Metab. 2018, 103, 4511–4523. [Google Scholar] [CrossRef]
- Pereira, S.S.; Monteiro, M.P.; Bourdeau, I.; Lacroix, A.; Pignatelli, D. Mechanisms of endocrinology: Cell cycle regulation in adrenocortical carcinoma. Eur. J. Endocrinol. 2018, 179, R95–R110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, W.; Wang, Q.; Zhu, Y.; Zhai, C.; Wang, J.; Yan, X.; Chai, L.; Li, M. Clinicopathological and prognostic significance of leucine-rich repeats and immunoglobulin-like domains protein 1 (LRIG1) in malignant tumors: A meta-analysis. J. Cancer 2018, 9, 2895–2909. [Google Scholar] [CrossRef]
- Torigoe, H.; Yamamoto, H.; Sakaguchi, M.; Youyi, C.; Namba, K.; Sato, H.; Shien, K.; Soh, J.; Suzawa, K.; Tomida, S.; et al. Tumor-suppressive effect of LRIG1, a negative regulator of ErbB, in non-small cell lung cancer harboring mutant EGFR. Carcinogenesis 2018, 39, 719–727. [Google Scholar] [CrossRef]
- Mao, F.; Wang, B.; Xiao, Q.; Cheng, F.; Lei, T.; Guo, D. LRIG proteins in glioma: Functional roles, molecular mechanisms, and potential clinical implications. J. Neurol. Sci. 2017, 383, 56–60. [Google Scholar] [CrossRef]
- Ragazzon, B.; Libe, R.; Gaujoux, S.; Assie, G.; Fratticci, A.; Launay, P.; Clauser, E.; Bertagna, X.; Tissier, F.; de Reynies, A.; et al. Transcriptome analysis reveals that p53 and {beta}-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res. 2010, 70, 8276–8281. [Google Scholar] [CrossRef] [Green Version]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagnere, A.M.; Rene-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, L.J.; Heinze, B.; Fassnacht, M.; Willenberg, H.S.; Quinkler, M.; Reisch, N.; Zink, M.; Allolio, B.; Hahner, S. TP53 germline mutations in adult patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E476–E485. [Google Scholar] [CrossRef] [Green Version]
- Raymond, V.M.; Else, T.; Everett, J.N.; Long, J.M.; Gruber, S.B.; Hammer, G.D. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E119–E125. [Google Scholar] [CrossRef] [Green Version]
- Giotti, B.; Chen, S.H.; Barnett, M.W.; Regan, T.; Ly, T.; Wiemann, S.; Hume, D.A.; Freeman, T.C. Assembly of a parts list of the human mitotic cell cycle machinery. J. Mol. Cell. Biol. 2019, 11, 703–718. [Google Scholar] [CrossRef]
- Ajioka, I. Coordination of proliferation and neuronal differentiation by the retinoblastoma protein family. Dev. Growth Differ. 2014, 56, 324–334. [Google Scholar] [CrossRef]
- Hardwick, L.J.; Ali, F.R.; Azzarelli, R.; Philpott, A. Cell cycle regulation of proliferation versus differentiation in the central nervous system. Cell Tissue Res. 2015, 359, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Vaudry, D.; Stork, P.J.; Lazarovici, P.; Eiden, L.E. Signaling pathways for PC12 cell differentiation: Making the right connections. Science 2002, 296, 1648–1649. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar]
- Jensch, A.; Thomaseth, C.; Radde, N.E. Sampling-based Bayesian approaches reveal the importance of quasi-bistable behavior in cellular decision processes on the example of the MAPK signaling pathway in PC-12 cell lines. BMC Syst. Biol. 2017, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Offermann, B.; Knauer, S.; Singh, A.; Fernandez-Cachon, M.L.; Klose, M.; Kowar, S.; Busch, H.; Boerries, M. Boolean Modeling Reveals the Necessity of Transcriptional Regulation for Bistability in PC12 Cell Differentiation. Front. Genet. 2016, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.D.; Verveer, P.J.; Bastiaens, P.I. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 2007, 9, 324–330. [Google Scholar] [CrossRef]
- Moriguchi, T.; Gotoh, Y.; Nishida, E. Activation of two isoforms of mitogen-activated protein kinase kinase in response to epidermal growth factor and nerve growth factor. Eur. J. Biochem. 1995, 234, 32–38. [Google Scholar]
- Tipping, A.J.; Pina, C.; Castor, A.; Hong, D.; Rodrigues, N.P.; Lazzari, L.; May, G.E.; Jacobsen, S.E.; Enver, T. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 2009, 113, 2661–2672. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E.; et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 2012, 149, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.; Vazquez, I.; Conchillo, A.; Garcia-Sanchez, M.A.; Marcotegui, N.; Fuster, O.; Gonzalez, M.; Calasanz, M.J.; Lahortiga, I.; Odero, M.D. Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia 2012, 26, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.; Conchillo, A.; Garcia-Sanchez, M.A.; Odero, M.D. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 2012, 82, 1–17. [Google Scholar] [CrossRef]
- Zheng, R.; Blobel, G.A. GATA Transcription Factors and Cancer. Genes Cancer 2010, 1, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranasic, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucl. Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Xu, X.; Bieda, M.; Jin, V.X.; Rabinovich, A.; Oberley, M.J.; Green, R.; Farnham, P.J. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007, 17, 1550–1561. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Fraenkel, E.; Pabo, C.O.; Pavletich, N.P. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 1999, 13, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.; Boyd, K.E.; Fry, C.J.; Bartley, S.M.; Farnham, P.J. Target gene specificity of E2F and pocket protein family members in living cells. Mol. Cell. Biol. 2000, 20, 5797–5807. [Google Scholar] [CrossRef] [Green Version]
- To, B.; Andrechek, E.R. Transcription factor compensation during mammary gland development in E2F knockout mice. PLoS ONE 2018, 13, e0194937. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.J.; Chang, J.T.; Bild, A.H.; Nevins, J.R. Compensation and specificity of function within the E2F family. Oncogene 2007, 26, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Vanderluit, J.L.; Ferguson, K.L.; Nikoletopoulou, V.; Parker, M.; Ruzhynsky, V.; Alexson, T.; McNamara, S.M.; Park, D.S.; Rudnicki, M.; Slack, R.S. p107 regulates neural precursor cells in the mammalian brain. J. Cell. Biol. 2004, 166, 853–863. [Google Scholar] [CrossRef]
- Vanderluit, J.L.; Wylie, C.A.; McClellan, K.A.; Ghanem, N.; Fortin, A.; Callaghan, S.; MacLaurin, J.G.; Park, D.S.; Slack, R.S. The Retinoblastoma family member p107 regulates the rate of progenitor commitment to a neuronal fate. J. Cell. Biol. 2007, 178, 129–139. [Google Scholar] [CrossRef]
- Ruzhynsky, V.A.; McClellan, K.A.; Vanderluit, J.L.; Jeong, Y.; Furimsky, M.; Park, D.S.; Epstein, D.J.; Wallace, V.A.; Slack, R.S. Cell cycle regulator E2F4 is essential for the development of the ventral telencephalon. J. Neurosci. 2007, 27, 5926–5935. [Google Scholar] [CrossRef]
- McClellan, K.A.; Ruzhynsky, V.A.; Douda, D.N.; Vanderluit, J.L.; Ferguson, K.L.; Chen, D.; Bremner, R.; Park, D.S.; Leone, G.; Slack, R.S. Unique requirement for Rb/E2F3 in neuronal migration: Evidence for cell cycle-independent functions. Mol. Cell. Biol. 2007, 27, 4825–4843. [Google Scholar] [CrossRef] [Green Version]
- McClellan, K.A.; Vanderluit, J.L.; Julian, L.M.; Andrusiak, M.G.; Dugal-Tessier, D.; Park, D.S.; Slack, R.S. The p107/E2F pathway regulates fibroblast growth factor 2 responsiveness in neural precursor cells. Mol. Cell. Biol. 2009, 29, 4701–4713. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.X.; Sheldrick, M.; Desbois, A.; Slinn, J.; Hou, S.T. Neuropilin-1 is a direct target of the transcription factor E2F1 during cerebral ischemia-induced neuronal death in vivo. Mol. Cell. Biol. 2007, 27, 1696–1705. [Google Scholar] [CrossRef] [Green Version]
- Andrusiak, M.G.; McClellan, K.A.; Dugal-Tessier, D.; Julian, L.M.; Rodrigues, S.P.; Park, D.S.; Kennedy, T.E.; Slack, R.S. Rb/E2F regulates expression of neogenin during neuronal migration. Mol. Cell. Biol. 2011, 31, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Ghanem, N.; Andrusiak, M.G.; Svoboda, D.; Al Lafi, S.M.; Julian, L.M.; McClellan, K.A.; De Repentigny, Y.; Kothary, R.; Ekker, M.; Blais, A.; et al. The Rb/E2F pathway modulates neurogenesis through direct regulation of the Dlx1/Dlx2 bigene cluster. J. Neurosci. 2012, 32, 8219–8230. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.M.; Vandenbosch, R.; Pakenham, C.A.; Andrusiak, M.G.; Nguyen, A.P.; McClellan, K.A.; Svoboda, D.S.; Lagace, D.C.; Park, D.S.; Leone, G.; et al. Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell 2013, 12, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.M.; Blais, A. Transcriptional control of stem cell fate by E2Fs and pocket proteins. Front. Genet. 2015, 6, 161. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.M.; Liu, Y.; Pakenham, C.A.; Dugal-Tessier, D.; Ruzhynsky, V.; Bae, S.; Tsai, S.Y.; Leone, G.; Slack, R.S.; Blais, A. Tissue-specific targeting of cell fate regulatory genes by E2f factors. Cell Death Differ. 2016, 23, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Sage, J. Novel functions for the transcription factor E2F4 in development and disease. Cell Cycle 2016, 15, 3183–3190. [Google Scholar] [CrossRef]
- Hsu, J.; Arand, J.; Chaikovsky, A.; Mooney, N.A.; Demeter, J.; Brison, C.M.; Oliverio, R.; Vogel, H.; Rubin, S.M.; Jackson, P.K.; et al. E2F4 regulates transcriptional activation in mouse embryonic stem cells independently of the RB family. Nat. Commun. 2019, 10, 2939. [Google Scholar] [CrossRef] [Green Version]
- Cuitino, M.C.; Pecot, T.; Sun, D.; Kladney, R.; Okano-Uchida, T.; Shinde, N.; Saeed, R.; Perez-Castro, A.J.; Webb, A.; Liu, T.; et al. Two Distinct E2F Transcriptional Modules Drive Cell Cycles and Differentiation. Cell Rep. 2019, 27, 3547–3560. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell. Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucl. Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Uxa, S.; Bernhart, S.H.; Mages, C.F.S.; Fischer, M.; Kohler, R.; Hoffmann, S.; Stadler, P.F.; Engeland, K.; Muller, G.A. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucl. Acids Res. 2019, 47, 9087–9103. [Google Scholar] [CrossRef] [Green Version]
- Schade, A.E.; Oser, M.G.; Nicholson, H.E.; DeCaprio, J.A. Cyclin D-CDK4 relieves cooperative repression of proliferation and cell cycle gene expression by DREAM and RB. Oncogene 2019, 38, 4962–4976. [Google Scholar] [CrossRef]
- Schade, A.E.; Fischer, M.; DeCaprio, J.A. RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucl. Acids Res. 2019, 47, 11197–11208. [Google Scholar] [CrossRef]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Sadasivam, S.; Duan, S.; DeCaprio, J.A. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012, 26, 474–489. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Muller, G.A. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef] [Green Version]
- Litovchick, L.; Sadasivam, S.; Florens, L.; Zhu, X.; Swanson, S.K.; Velmurugan, S.; Chen, R.; Washburn, M.P.; Liu, X.S.; DeCaprio, J.A. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 2007, 26, 539–551. [Google Scholar] [CrossRef]
- Zhang, W.; Tyl, M.; Ward, R.; Sobott, F.; Maman, J.; Murthy, A.S.; Watson, A.A.; Fedorov, O.; Bowman, A.; Owen-Hughes, T.; et al. Structural plasticity of histones H3-H4 facilitates their allosteric exchange between RbAp48 and ASF1. Nat. Struct. Mol. Biol. 2013, 20, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Murzina, N.V.; Pei, X.Y.; Zhang, W.; Sparkes, M.; Vicente-Garcia, J.; Pratap, J.V.; McLaughlin, S.H.; Ben-Shahar, T.R.; Verreault, A.; Luisi, B.F.; et al. Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure 2008, 16, 1077–1085. [Google Scholar] [CrossRef]
- Qian, Y.W.; Wang, Y.C.; Hollingsworth, R.E., Jr.; Jones, D.; Ling, N.; Lee, E.Y. A retinoblastoma-binding protein related to a negative regulator of Ras in yeast. Nature 1993, 364, 648–652. [Google Scholar] [CrossRef]
- Kitamura, H.; Ozono, E.; Iwanaga, R.; Bradford, A.P.; Okuno, J.; Shimizu, E.; Kurayoshi, K.; Kugawa, K.; Toh, H.; Ohtani, K. Identification of novel target genes specifically activated by deregulated E2F in human normal fibroblasts. Genes Cells 2015, 20, 739–757. [Google Scholar] [CrossRef] [Green Version]
- Schmit, F.; Cremer, S.; Gaubatz, S. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J. 2009, 276, 5703–5716. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, S.; Siu, T.L.; Huang, S. Glioblastoma multiforme formation and EMT: Role of FoxM1 transcription factor. Curr. Pharm. Des. 2015, 21, 1268–1271. [Google Scholar] [CrossRef] [Green Version]
- Coulson, J.M. Transcriptional regulation: Cancer, neurons and the REST. Curr. Biol. 2005, 15, R665–R668. [Google Scholar] [CrossRef] [Green Version]
- Lunyak, V.V.; Rosenfeld, M.G. No rest for REST: REST/NRSF regulation of neurogenesis. Cell 2005, 121, 499–501. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Prada, I.; D’Alessandro, R.; Meldolesi, J. REST: An oncogene or a tumor suppressor? Trends Cell. Biol. 2013, 23, 289–295. [Google Scholar] [CrossRef]
- Li, C.; Wang, Z.; Tang, X.; Zeng, L.; Fan, X.; Li, Z. Molecular mechanisms and potential prognostic effects of REST and REST4 in glioma (Review). Mol. Med. Rep. 2017, 16, 3707–3712. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhu, M.; Yu, Y.; Qiu, L.; Zhang, Y.; He, L.; Zhang, J. Brain REST/NRSF Is Not Only a Silent Repressor but Also an Active Protector. Mol. Neurobiol. 2017, 54, 541–550. [Google Scholar] [CrossRef]
- Ren, X.; Kerppola, T.K. REST interacts with Cbx proteins and regulates polycomb repressive complex 1 occupancy at RE1 elements. Mol. Cell. Biol. 2011, 31, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Niklison-Chirou, M.V.; Agostini, M.; Amelio, I.; Melino, G. Regulation of Adult Neurogenesis in Mammalian Brain. Int. J. Mol. Sci. 2020, 21, 4869. [Google Scholar] [CrossRef]
- Mozzi, A.; Guerini, F.R.; Forni, D.; Costa, A.S.; Nemni, R.; Baglio, F.; Cabinio, M.; Riva, S.; Pontremoli, C.; Clerici, M.; et al. REST, a master regulator of neurogenesis, evolved under strong positive selection in humans and in non human primates. Sci. Rep. 2017, 7, 9530. [Google Scholar] [CrossRef]
- Urban, N.; Blomfield, I.M.; Guillemot, F. Quiescence of Adult Mammalian Neural Stem Cells: A Highly Regulated Rest. Neuron 2019, 104, 834–848. [Google Scholar] [CrossRef]
- Chen, G.L.; Miller, G.M. Alternative REST Splicing Underappreciated. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Shafik, A.; Schumann, U.; Evers, M.; Sibbritt, T.; Preiss, T. The emerging epitranscriptomics of long noncoding RNAs. Biochim. Biophys. Acta 2016, 1859, 59–70. [Google Scholar] [CrossRef]
- Bonasio, R.; Shiekhattar, R. Regulation of transcription by long noncoding RNAs. Annu. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef] [Green Version]
- Yan, P.; Luo, S.; Lu, J.Y.; Shen, X. Cis- and trans-acting lncRNAs in pluripotency and reprogramming. Curr. Opin. Genet. Dev. 2017, 46, 170–178. [Google Scholar] [CrossRef]
- Hansji, H.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Keeping abreast with long non-coding RNAs in mammary gland development and breast cancer. Front. Genet. 2014, 5, 379. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Wang, M.; Ma, N.; Xu, Y.; Jiang, Y.; Gao, X. Long noncoding RNAs: Novel players in colorectal cancer. Cancer Lett. 2015, 361, 13–21. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Wang, J.; Song, Y.; Gao, P.; Shi, J.; Chen, P.; Wang, Z. Long noncoding RNAs in gastric cancer: Functions and clinical applications. Onco Targets Ther. 2016, 9, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Pastori, C.; Wahlestedt, C. Involvement of long noncoding RNAs in diseases affecting the central nervous system. RNA Biol. 2012, 9, 860–870. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.; Setia, M.; Kaur, N.; Shepal, V.; Arora, V.; Singh, D.K.; Mondal, A.; Teli, A.; Tathode, M.; Gajula, R.; et al. Noncoding RNA Ginir functions as an oncogene by associating with centrosomal proteins. PLoS Biol. 2018, 16, e2004204. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wei, Y.; Yan, X.; Li, N.; Song, H.; Yang, L.; Wu, Y.; Xi, Y.F.; Weng, H.W.; Li, J.H.; et al. Survivin is a prognostic marker and therapeutic target for extranodal, nasal-type natural killer/T cell lymphoma. Ann. Transl. Med. 2019, 7, 316. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin—The inconvenient IAP. Semin. Cell Dev. Biol. 2015, 39, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Preusser, M.; Gelpi, E.; Matej, R.; Marosi, C.; Dieckmann, K.; Rossler, K.; Budka, H.; Hainfellner, J.A. No prognostic impact of survivin expression in glioblastoma. Acta Neuropathol. 2005, 109, 534–538. [Google Scholar] [CrossRef]
- Tong, X.; Yang, P.; Wang, K.; Liu, Y.; Liu, X.; Shan, X.; Huang, R.; Zhang, K.; Wang, J. Survivin is a prognostic indicator in glioblastoma and may be a target of microRNA-218. Oncol. Lett 2019, 18, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Farmer, G.; Friedlander, P.; Colgan, J.; Manley, J.L.; Prives, C. Transcriptional repression by p53 involves molecular interactions distinct from those with the TATA box binding protein. Nucl. Acids Res. 1996, 24, 4281–4288. [Google Scholar] [CrossRef] [Green Version]
- Truant, R.; Xiao, H.; Ingles, C.J.; Greenblatt, J. Direct interaction between the transcriptional activation domain of human p53 and the TATA box-binding protein. J. Biol. Chem. 1993, 268, 2284–2287. [Google Scholar]
- St Clair, S.; Giono, L.; Varmeh-Ziaie, S.; Resnick-Silverman, L.; Liu, W.J.; Padi, A.; Dastidar, J.; DaCosta, A.; Mattia, M.; Manfredi, J.J. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: One involves direct binding to the cdc25C promoter. Mol. Cell 2004, 16, 725–736. [Google Scholar] [CrossRef]
- Giono, L.E.; Resnick-Silverman, L.; Carvajal, L.A.; St Clair, S.; Manfredi, J.J. Mdm2 promotes Cdc25C protein degradation and delays cell cycle progression through the G2/M phase. Oncogene 2017, 36, 6762–6773. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lu, R.; Zhao, Q.; Du, J.; Li, Y.; Zheng, M.; Zhang, S. Association and clinicopathologic significance of p38MAPK-ERK-JNK-CDC25C with polyploid giant cancer cell formation. Med. Oncol. 2019, 37, 6. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget 2017, 8, 624–643. [Google Scholar] [CrossRef] [Green Version]
- Musa, J.; Aynaud, M.M.; Mirabeau, O.; Delattre, O.; Grunewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef]
- Bayley, R.; Ward, C.; Garcia, P. MYBL2 amplification in breast cancer: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188407. [Google Scholar] [CrossRef]
- Sant, K.E.; Nahar, M.S.; Dolinoy, D.C. DNA methylation screening and analysis. Methods Mol. Biol 2012, 889, 385–406. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.Y.; Meaney, M.J. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 2010, 61, 439–466. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Fan, G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 2009, 89, 67–84. [Google Scholar] [CrossRef]
- Rauscher, G.H.; Kresovich, J.K.; Poulin, M.; Yan, L.; Macias, V.; Mahmoud, A.M.; Al-Alem, U.; Kajdacsy-Balla, A.; Wiley, E.L.; Tonetti, D.; et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer 2015, 15, 816. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Guez-Haddad, J.; Sporny, M.; Sasson, Y.; Gevorkyan-Airapetov, L.; Lahav-Mankovski, N.; Margulies, D.; Radzimanowski, J.; Opatowsky, Y. The Neuronal Migration Factor srGAP2 Achieves Specificity in Ligand Binding through a Two-Component Molecular Mechanism. Structure 2015, 23, 1989–2000. [Google Scholar] [CrossRef] [Green Version]
- Fritz, R.D.; Menshykau, D.; Martin, K.; Reimann, A.; Pontelli, V.; Pertz, O. SrGAP2-Dependent Integration of Membrane Geometry and Slit-Robo-Repulsive Cues Regulates Fibroblast Contact Inhibition of Locomotion. Dev. Cell 2015, 35, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Yuan, Q.; Braun, M.; Zhang, X.; Petri, B.; Zhang, J.; Kim, D.; Guez-Haddad, J.; Xue, W.; Pan, W.; et al. Leukocyte Cytoskeleton Polarization Is Initiated by Plasma Membrane Curvature from Cell Attachment. Dev. Cell 2019, 49, 206–219.e7. [Google Scholar] [CrossRef]
- Mason, F.M.; Heimsath, E.G.; Higgs, H.N.; Soderling, S.H. Bi-modal regulation of a formin by srGAP2. J. Biol. Chem. 2011, 286, 6577–6586. [Google Scholar] [CrossRef] [Green Version]
- Kan, S.; Chai, S.; Chen, W.; Yu, B. DNA methylation profiling identifies potentially significant epigenetically-regulated genes in glioblastoma multiforme. Oncol. Lett. 2019, 18, 1679–1688. [Google Scholar] [CrossRef]
- Diez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenet. Chromatin 2015, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Munzel, M.; Globisch, D.; Bruckl, T.; Wagner, M.; Welzmiller, V.; Michalakis, S.; Muller, M.; Biel, M.; Carell, T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew. Chem. Int. Ed. Engl. 2010, 49, 5375–5377. [Google Scholar] [CrossRef]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef] [Green Version]
- Stricker, S.H.; Gotz, M. DNA-Methylation: Master or Slave of Neural Fate Decisions? Front. Neurosci. 2018, 12, 5. [Google Scholar] [CrossRef]
- Noack, F.; Pataskar, A.; Schneider, M.; Buchholz, F.; Tiwari, V.K.; Calegari, F. Assessment and site-specific manipulation of DNA (hydroxy-)methylation during mouse corticogenesis. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Santiago, M.; Antunes, C.; Guedes, M.; Sousa, N.; Marques, C.J. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, M.J.; Wojtas, B. Global DNA Methylation Patterns in Human Gliomas and Their Interplay with Other Epigenetic Modifications. Int. J. Mol. Sci. 2019, 20, 3478. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef] [Green Version]
- Cochran, J.N.; Geier, E.G.; Bonham, L.W.; Newberry, J.S.; Amaral, M.D.; Thompson, M.L.; Lasseigne, B.N.; Karydas, A.M.; Roberson, E.D.; Cooper, G.M.; et al. Non-coding and Loss-of-Function Coding Variants in TET2 are Associated with Multiple Neurodegenerative Diseases. Am. J. Hum. Genet. 2020, 106, 632–645. [Google Scholar] [CrossRef]
- Tremblay, M.; Sanchez-Ferras, O.; Bouchard, M. GATA transcription factors in development and disease. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.K.; Wang, Y.C. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M.; et al. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Koizumi, K. Family with sequence similarity 107: A family of stress responsive small proteins with diverse functions in cancer and the nervous system (Review). Biomed. Rep. 2014, 2, 321–325. [Google Scholar] [CrossRef]
- Ma, Y.S.; Wu, Z.J.; Bai, R.Z.; Dong, H.; Xie, B.X.; Wu, X.H.; Hang, X.S.; Liu, A.N.; Jiang, X.H.; Wang, G.R.; et al. DRR1 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via regulating AKT activation. Cancer Lett. 2018, 423, 86–94. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martinez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucl. Acids 2018, 11, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Yamato, T.; Orikasa, K.; Fukushige, S.; Orikasa, S.; Horii, A. Isolation and characterization of the novel gene, TU3A, in a commonly deleted region on 3p14.3-->p14.2 in renal cell carcinoma. Cytogenet. Cell Genet. 1999, 87, 291–295. [Google Scholar] [CrossRef]
- Wang, L.; Darling, J.; Zhang, J.S.; Liu, W.; Qian, J.; Bostwick, D.; Hartmann, L.; Jenkins, R.; Bardenhauer, W.; Schutte, J.; et al. Loss of expression of the DRR 1 gene at chromosomal segment 3p21.1 in renal cell carcinoma. Genes Chromosomes Cancer 2000, 27, 1–10. [Google Scholar] [CrossRef]
- De Rubis, G.; Rajeev Krishnan, S.; Bebawy, M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186. [Google Scholar] [CrossRef]
- Heese, K. The protein p17 signaling pathways in cancer. Tumour Biol. 2013, 34, 4081–4087. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Fang, L.; Cheng, Q.; Li, W.; Liu, J.; Li, L.; Xu, K.; Zheng, J. Antitumor activities of an oncolytic adenovirus equipped with a double siRNA targeting Ki67 and hTERT in renal cancer cells. Virus Res. 2014, 181, 61–71. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, M.; Xu, K.; Mao, L.; Zheng, J. shRNA-armed conditionally replicative adenoviruses: A promising approach for cancer therapy. Oncotarget 2016, 7, 29824–29834. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Castanotto, D.; Stein, C.A. Antisense oligonucleotides in cancer. Curr. Opin. Oncol. 2014, 26, 584–589. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.; Ding, M.; Xu, K.; Li, L.; Mao, L.; Zheng, J. Ki67 targeted strategies for cancer therapy. Clin. Transl. Oncol. 2018, 20, 570–575. [Google Scholar] [CrossRef]
- Kausch, I.; Lingnau, A.; Endl, E.; Sellmann, K.; Deinert, I.; Ratliff, T.L.; Jocham, D.; Sczakiel, G.; Gerdes, J.; Bohle, A. Antisense treatment against Ki-67 mRNA inhibits proliferation and tumor growth in vitro and in vivo. Int. J. Cancer 2003, 105, 710–716. [Google Scholar] [CrossRef]
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of targeting transforming growth factor beta-2 with antisense OT-101 on the cytokine and chemokine profile in patients with advanced pancreatic cancer. Onco Targets Ther. 2018, 11, 2779–2796. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [Green Version]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784. [Google Scholar] [CrossRef] [Green Version]
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3331. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ma, S.; Liu, Y.; Lu, W.; Sun, L.; Zhao, P.; Xia, Q. Transcriptional repression of endogenous genes in BmE cells using CRISPRi system. Insect Biochem. Mol. Biol. 2019, 111, 103172. [Google Scholar] [CrossRef]
- MacLeod, R.S.; Cawley, K.M.; Gubrij, I.; Nookaew, I.; Onal, M.; O’Brien, C.A. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci. Rep. 2019, 9, 17312. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell. Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, S.; Kutzner, A.; Heese, K. Lead discovery and in silico 3D structure modeling of tumorigenic FAM72A (p17). Tumour Biol. 2015, 36, 239–249. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. In Methods in Molecular Biology; Springer: Berlin, Germany, 2017; Volume 1520, pp. 85–106. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef]
- Pramanik, S.; Roy, K. Predictive modeling of chemical toxicity towards Pseudokirchneriella subcapitata using regression and classification based approaches. Ecotoxicol. Environ. Saf. 2014, 101, 184–190. [Google Scholar] [CrossRef]
- Pramanik, S.; Roy, K. Exploring QSTR modeling and toxicophore mapping for identification of important molecular features contributing to the chemical toxicity in Escherichia coli. Toxicol. In Vitro 2014, 28, 265–272. [Google Scholar] [CrossRef]
- van Montfort, R.L.; Workman, P. Structure-based design of molecular cancer therapeutics. Trends Biotechnol. 2009, 27, 315–328. [Google Scholar] [CrossRef]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucl. Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, N.T.T.; Rahane, C.S.; Pramanik, S.; Kim, P.-S.; Kutzner, A.; Heese, K. FAM72, Glioblastoma Multiforme (GBM) and Beyond. Cancers 2021, 13, 1025. https://doi.org/10.3390/cancers13051025
Ho NTT, Rahane CS, Pramanik S, Kim P-S, Kutzner A, Heese K. FAM72, Glioblastoma Multiforme (GBM) and Beyond. Cancers. 2021; 13(5):1025. https://doi.org/10.3390/cancers13051025
Chicago/Turabian StyleHo, Nguyen Thi Thanh, Chinmay Satish Rahane, Subrata Pramanik, Pok-Son Kim, Arne Kutzner, and Klaus Heese. 2021. "FAM72, Glioblastoma Multiforme (GBM) and Beyond" Cancers 13, no. 5: 1025. https://doi.org/10.3390/cancers13051025