
Altertoxin II, a Highly Effective and Specific Compound against Ewing Sarcoma

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
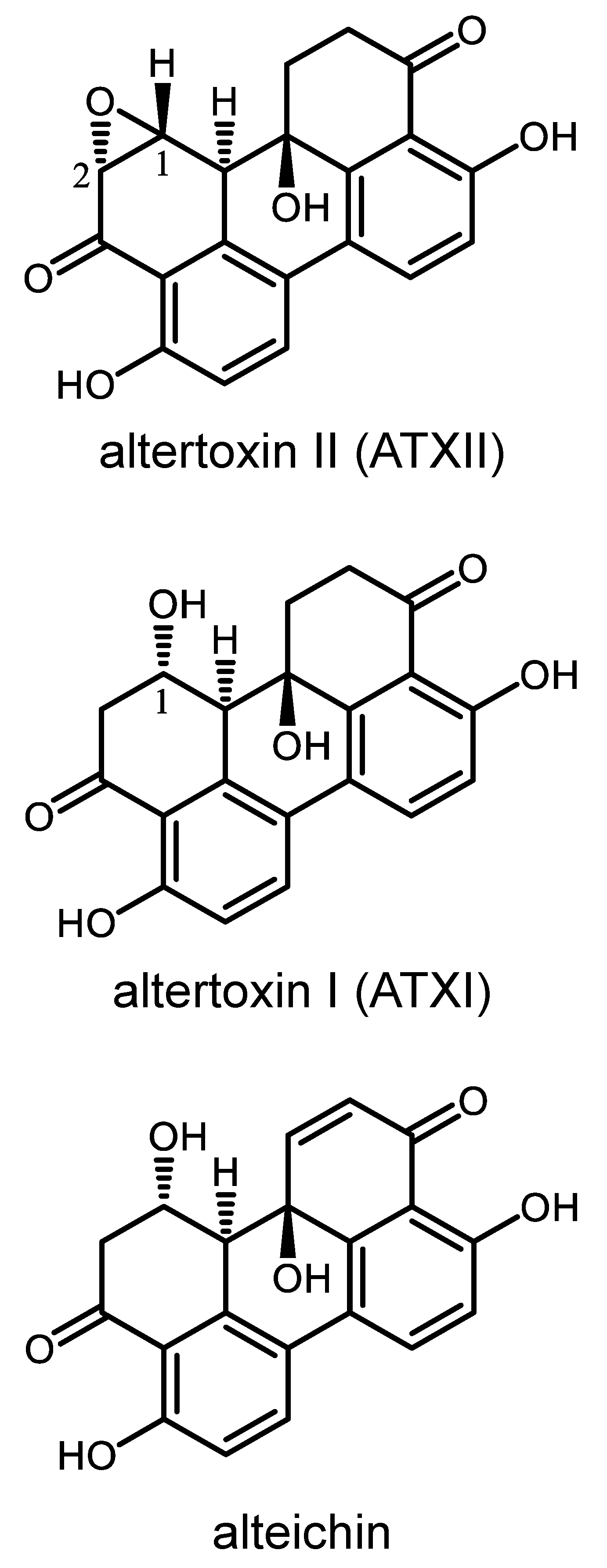
2.2. Purification of Secondary Metabolites Altertoxin II, Altertoxin I, and Alteichin
2.3. Phylogenetic Analysis of Citizen Science Alternaria
2.4. Preparation of Alternaria Isolates for Metabolomic Analysis
2.5. LCMS Detection of Altertoxin II
2.6. Cell Culture
2.7. Sulforhodamine B Assay
2.8. Colony Formation Assays
2.9. Whole-Cell Lysis and Immunoblotting
2.10. Flow Cytometry
2.11. RNA Sequencing
2.12. High-Content Immunofluorescence Imaging
2.13. Luciferase Reporter Assay
2.14. LLAMAS Assay
2.15. BioLayer Interferometry
2.16. Xenograft Studies in Nude Mice
3. Results
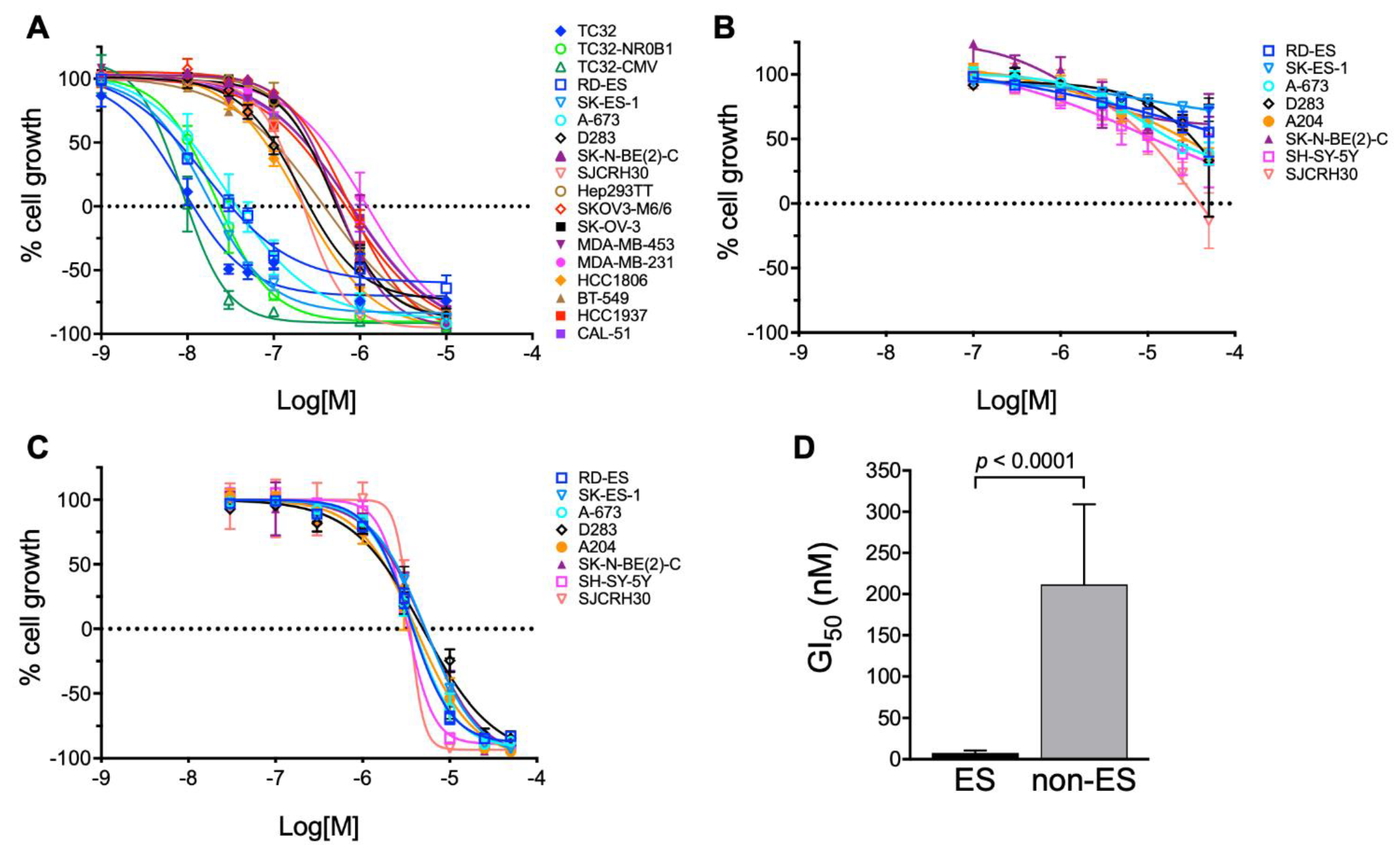
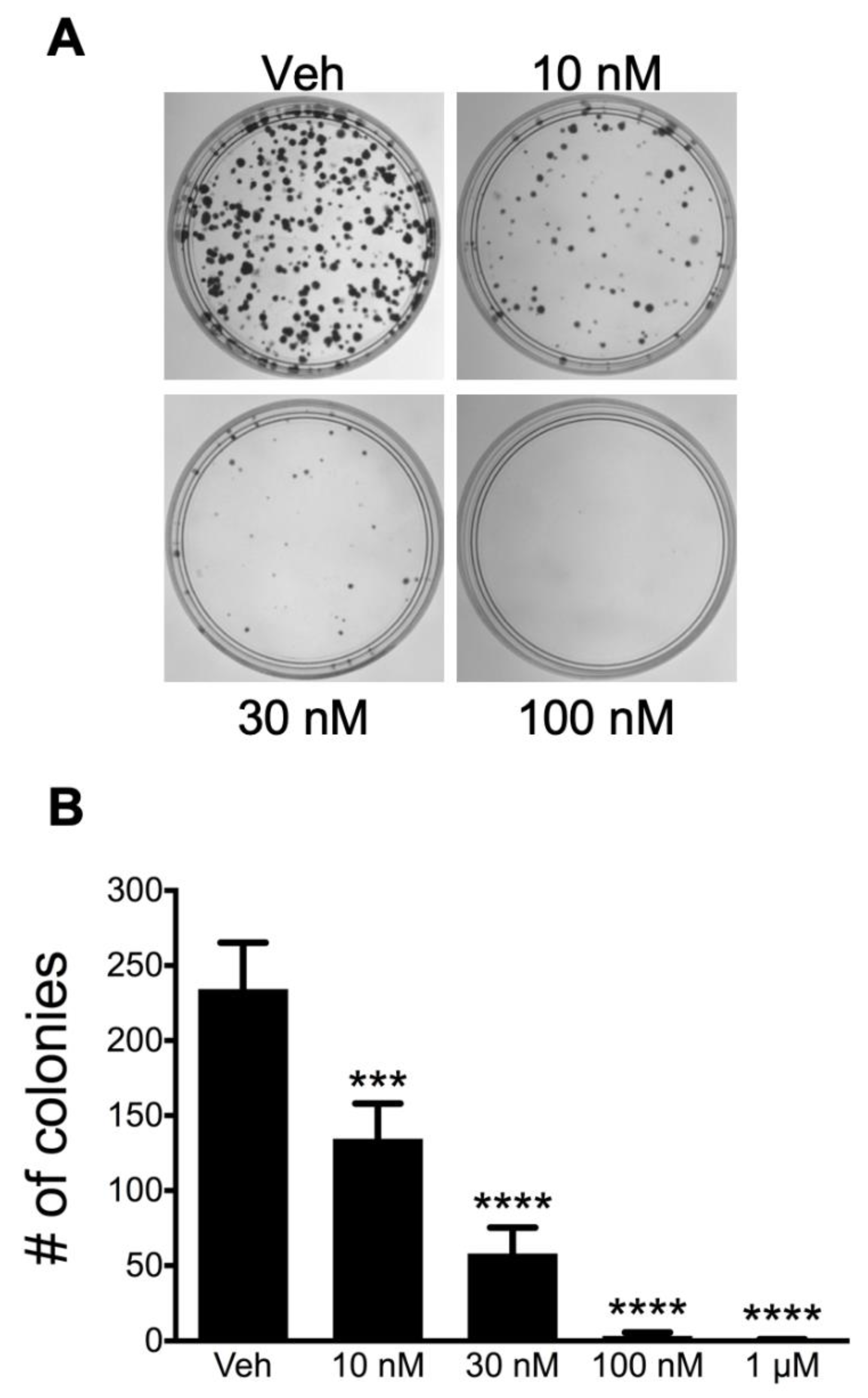
3.1. Bioassay-Guided Purification of Altertoxin II
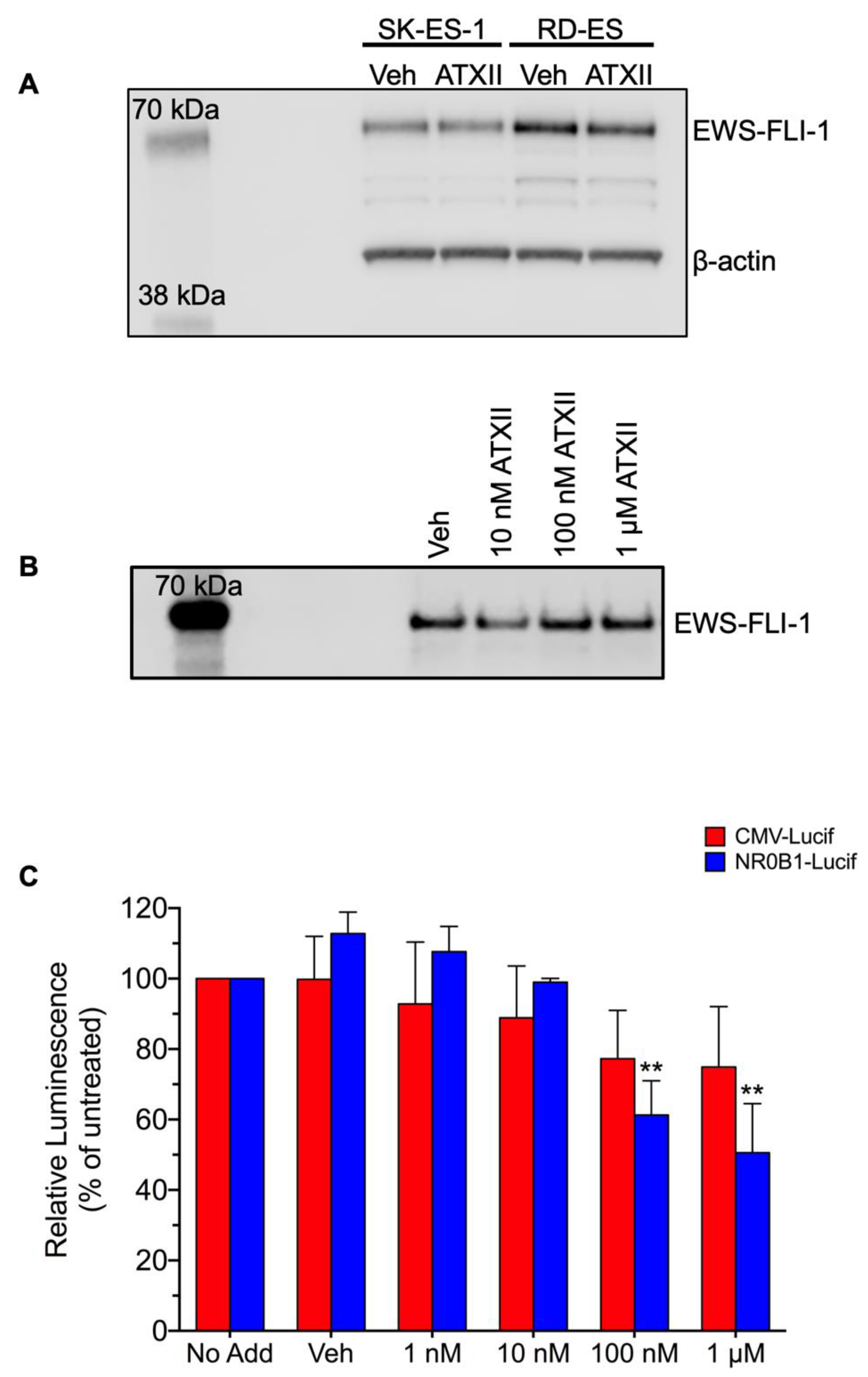
3.2. ATXII Does Not Inhibit EWS-FLI1 Protein Expression or Transcriptional Activity
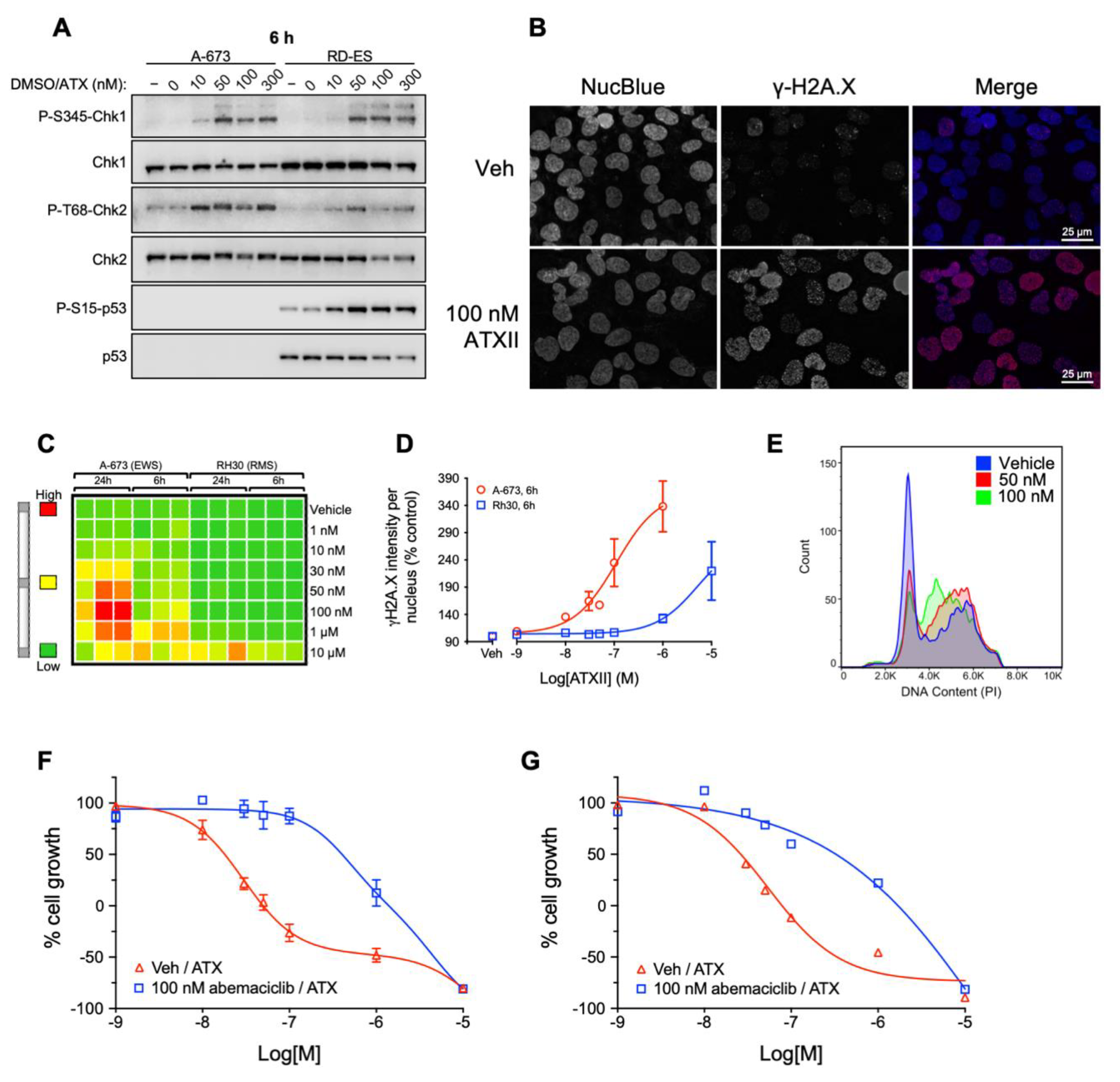
3.3. ATXII Activates DNA Damage Response Pathways and Induces Double-Strand DNA Breaks in ES Cells
3.4. ATXII Does Not Directly Bind to DNA
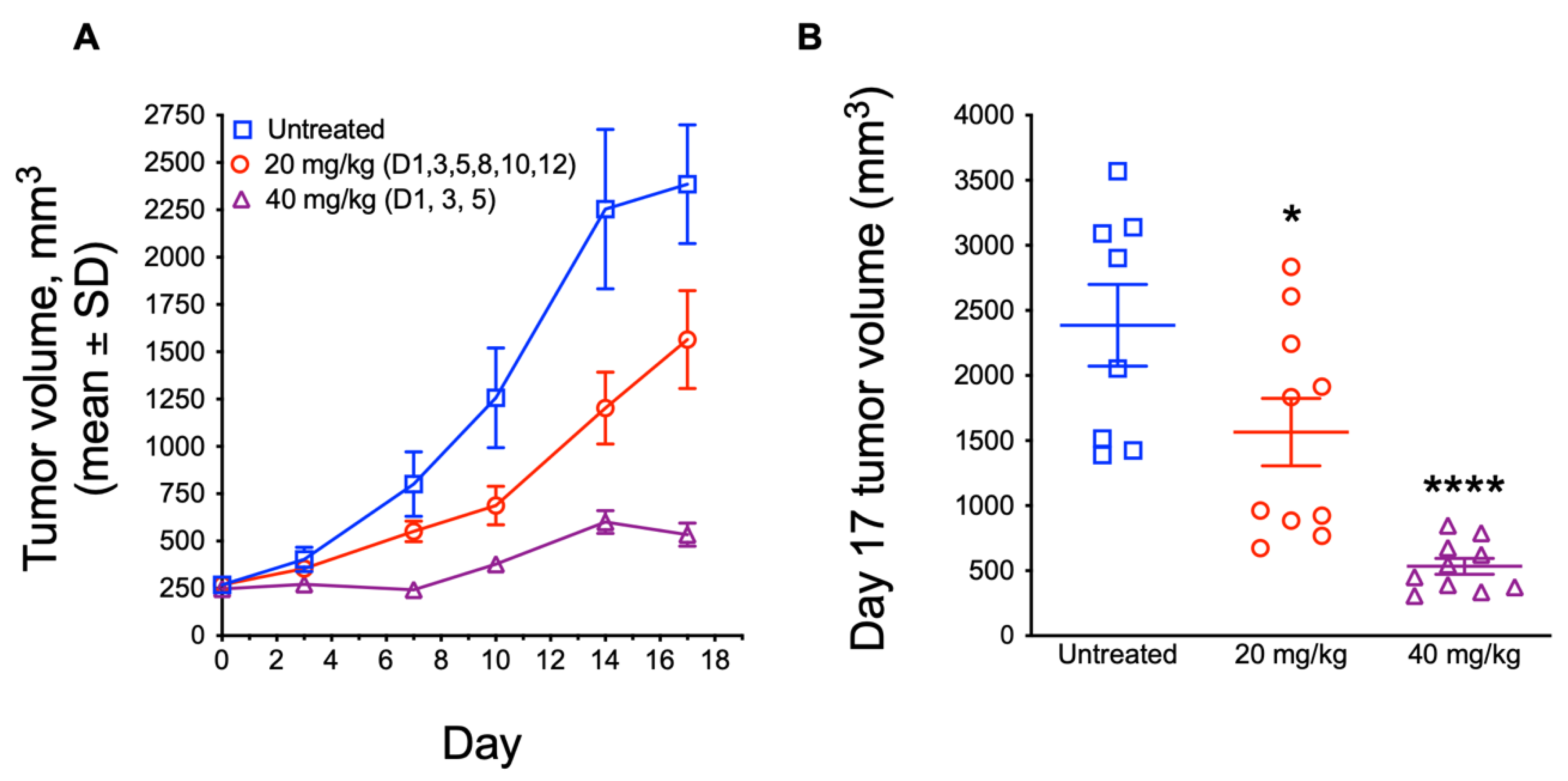
3.5. In Vivo Antitumor Efficacy of ATXII
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ge, Y.; Guo, L.; Huang, L. Potential approaches to the treatment of Ewing’s sarcoma. Oncotarget 2017, 8, 5523–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Üren, A.; Tcherkasskaya, O.; Toretsky, J.A. Recombinant EWS-FLI1 oncoprotein activates transcription. Biochemistry 2004, 43, 13579–13589. [Google Scholar] [CrossRef]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.-R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.-R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caropreso, V.; Darvishi, E.; Turbyville, T.J.; Ratnayake, R.; Grohar, P.J.; McMahon, J.B.; Woldemichael, G.M. Englerin A inhibits EWS-FLI1 DNA binding in Ewing sarcoma cells. J. Biol. Chem. 2016, 291, 10058–10066. [Google Scholar] [CrossRef] [Green Version]
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS-FLI1 fusion transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; Lee-Scott, M.; Adamson, P.C.; Blaney, S.M. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: A report from the Children’s Oncology Group. Eur. J. Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.J.; Cai, S.; Cichewicz, R.H.; Mooberry, S.L. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res. Treat. 2016, 157, 475–488. [Google Scholar] [CrossRef]
- Robles, A.J.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Maximiscin Induces DNA Damage, Activates DNA Damage Response Pathways, and Has Selective Cytotoxic Activity against a Subtype of Triple-Negative Breast Cancer. J. Nat. Prod. 2016, 79, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.J.; McCowen, S.; Cai, S.; Glassman, M.; Ruiz, F.; Cichewicz, R.H.; McHardy, S.F.; Mooberry, S.L. Structure-Activity Relationships of New Natural Product-Based Diaryloxazoles with Selective Activity against Androgen Receptor-Positive Breast Cancer Cells. J. Med. Chem. 2017, 60, 9275–9289. [Google Scholar] [CrossRef]
- Grant, C.V.; Carver, C.M.; Hastings, S.D.; Ramachandran, K.; Muniswamy, M.; Risinger, A.L.; Beutler, J.A.; Mooberry, S.L. Triple-negative breast cancer cell line sensitivity to englerin A identifies a new, targetable subtype. Breast Cancer Res. Treat. 2019, 177, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.S.; Risinger, A.L.; Petersen, C.L.; Mooberry, S.L.; Cichewicz, R.H. Leucinostatins from Ophiocordyceps spp. and Purpureocillium spp. Demonstrate Selective Antiproliferative Effects in Cells Representing the Luminal Androgen Receptor Subtype of Triple Negative Breast Cancer. J. Nat. Prod. 2020, 83, 2010–2024. [Google Scholar] [CrossRef]
- Pederson, P.J.; Cai, S.; Carver, C.; Powell, D.R.; Risinger, A.L.; Grkovic, T.; O’Keefe, B.R.; Mooberry, S.L.; Cichewicz, R.H. Triple-Negative Breast Cancer Cells Exhibit Differential Sensitivity to Cardenolides from Calotropis gigantea. J. Nat. Prod. 2020, 83, 2269–2280. [Google Scholar] [CrossRef]
- Cai, S.; Risinger, A.L.; Petersen, C.L.; Grkovic, T.; O’Keefe, B.R.; Mooberry, S.L.; Cichewicz, R.H. Anacolosins A–F and Corymbulosins X and Y, Clerodane Diterpenes from Anacolosa clarkii Exhibiting Cytotoxicity toward Pediatric Cancer Cell Lines. J. Nat. Prod. 2019, 82, 928–936. [Google Scholar] [CrossRef]
- Carter, A.C.; Petersen, C.L.; Wendt, K.L.; Helff, S.K.; Risinger, A.L.; Mooberry, S.L.; Cichewicz, R.H. In Situ Ring Contraction and Transformation of the Rhizoxin Macrocycle through an Abiotic Pathway. J. Nat. Prod. 2019, 82, 886–894. [Google Scholar] [CrossRef]
- Tinley, T.L.; Randall-Hlubek, D.A.; Leal, R.M.; Jackson, E.M.; Cessac, J.W.; Quada, J.C.; Hemscheidt, T.K.; Mooberry, S.L. Taccalonolides E and A: Plant-derived steroids with microtubule-stabilizing activity. Cancer Res. 2003, 63, 3211–3220. [Google Scholar] [PubMed]
- Risinger, A.L.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res. 2013, 73, 6780–6792. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Krishan, A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 1975, 66, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Overacker, R.D.; Plitzko, B.; Loesgen, S. Biolayer interferometry provides a robust method for detecting DNA binding small molecules in microbial extracts. Anal. Bioanal. Chem. 2021, 413, 1159–1171. [Google Scholar] [CrossRef]
- Shah, N.B.; Duncan, T.M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J. Vis. Exp. 2014, 84, e51383. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.; Tanious, F.A.; Wilson, W.D. Biosensor-surface plasmon resonance: Quantitative analysis of small molecule-nucleic acid interactions. Methods 2007, 42, 150–161. [Google Scholar] [CrossRef]
- Wahl, H.E.; Raudabaugh, D.B.; Bach, E.M.; Bone, T.S.; Luttenton, M.R.; Cichewicz, R.H.; Miller, A.N. What lies beneath? Fungal diversity at the bottom of Lake Michigan and Lake Superior. J. Great Lakes Res. 2018, 44, 263–270. [Google Scholar] [CrossRef]
- Du, L.; Robles, A.J.; King, J.B.; Powell, D.R.; Miller, A.N.; Mooberry, S.L.; Cichewicz, R.H. Crowdsourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. Int. Ed. Engl. 2014, 53, 804–809. [Google Scholar] [CrossRef] [Green Version]
- Hradil, C.M.; Hallock, Y.F.; Clardy, J.; Kenfield, D.S.; Strobel, G. Phytotoxins from Alternaria cassiae. Phytochemistry 1989, 28, 73–75. [Google Scholar] [CrossRef]
- Arnone, A.; Nasini, G.; Merlini, L.; Assante, G. Secondary mould metabolites. Part 16. Stemphyltoxins, new reduced perylenequinone metabolites from Stemphylium botryosum var. lactucum. J. Chem. Soc. Perkin Trans. 1986, 1, 525–530. [Google Scholar] [CrossRef]
- Stack, M.E.; Mazzola, E.P.; Page, S.W.; Pohland, A.E.; Highet, R.J.; Tempesta, M.S.; Corley, D.G. Mutagenic Perylenequinone Metabolites of Alternaria Alternata: Altertoxins I., II, and III. J. Nat. Prod. 1986, 49, 866–871. [Google Scholar] [CrossRef]
- Okuno, T.; Natsume, I.; Sawai, K.; Sawamura, K.; Furusaki, A.; Matsumoto, T. Structure of antifungal and phytotoxic pigments produced by alternaria sps. Tetrahedron Lett. 1983, 24, 5653–5656. [Google Scholar] [CrossRef]
- Podlech, J.; Fleck, S.C.; Metzler, M.; Bürck, J.; Ulrich, A.S. Determination of the Absolute Configuration of Perylene Quinone-Derived Mycotoxins by Measurement and Calculation of Electronic Circular Dichroism Spectra and Specific Rotations. Chem. A Eur. J. 2014, 20, 11463–11470. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Melot, T.; Garau, X.S.; Zucker, J.-M.; Lenoir, G.M.; Ambros, P.F.; Sheer, D.; Turc-Carel, C.; Triche, T.J.; et al. The Ewing Family of Tumors—A Subgroup of Small-Round-Cell Tumors Defined by Specific Chimeric Transcripts. N. Engl. J. Med. 1994, 331, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Bailly, R.A.; Bosselut, R.; Zucman, J.; Cormier, F.; Delattre, O.; Roussel, M.; Thomas, G.; Ghysdael, J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol. Cell. Biol. 1994, 14, 3230–3241. [Google Scholar] [CrossRef] [Green Version]
- May, W.A.; Gishizky, M.L.; Lessnick, S.L.; Lunsford, L.B.; Lewis, B.C.; Delattre, O.; Zucman, J.; Thomas, G.; Denny, C.T. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 5752–5756. [Google Scholar] [CrossRef] [Green Version]
- Stack, M.E.; Prival, M.J. Mutagenicity of the Alternaria metabolites altertoxins I, II, and III. Appl. Environ. Microbiol. 1986, 52, 718–722. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, C.; Tiessen, C.; Kreutzer, M.; Stark, T.; Hofmann, T.; Marko, D. Characterization of a genotoxic impact compound in alternaria alternata infested rice as altertoxin II. Arch. Toxicol. 2012, 86, 1911–1925. [Google Scholar] [CrossRef]
- Fleck, S.C.; Burkhardt, B.; Pfeiffer, E.; Metzler, M. Alternaria toxins: Altertoxin II is a much stronger mutagen and DNA strand breaking mycotoxin than alternariol and its methyl ether in cultured mammalian cells. Toxicol. Lett. 2012, 214, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS–FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef] [PubMed]
- May, W.A.; Grigoryan, R.S.; Keshelava, N.; Cabral, D.J.; Christensen, L.L.; Jenabi, J.; Ji, L.; Triche, T.J.; Lawlor, E.R.; Reynolds, C.P. Characterization and Drug Resistance Patterns of Ewing’s Sarcoma Family Tumor Cell Lines. PLoS ONE 2013, 8, e80060. [Google Scholar] [CrossRef] [PubMed]
- Benedict, W.F.; Baker, M.S.; Haroun, L.; Choi, E.; Ames, B.N. Mutagenicity of cancer chemotherapeutic agents in the Salmonella/microsome test. Cancer Res. 1977, 37, 2209–2213. [Google Scholar]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Soler, M.; Morgado-Palacin, I.; Lafarga, V.; Lecona, E.; Murga, M.; Callen, E.; Azorin, D.; Alonso, J.; Lopez-Contreras, A.J.; Nussenzweig, A.; et al. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget 2016, 7, 58759–58767. [Google Scholar] [CrossRef] [Green Version]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liang, H.; Cai, S.; O’Keefe, B.R.; Mooberry, S.L.; Cichewicz, R.H. An Integrated Strategy for the Detection, Dereplication, and Identification of DNA-Binding Biomolecules from Complex Natural Product Mixtures. J. Nat. Prod. 2020, 84, 750–761. [Google Scholar] [CrossRef]
- Marina, N.M.; Liu, Q.; Donaldson, S.S.; Sklar, C.A.; Armstrong, G.T.; Oeffinger, K.C.; Leisenring, W.M.; Ginsberg, J.P.; Henderson, T.O.; Neglia, J.P.; et al. Longitudinal follow-up of adult survivors of Ewing sarcoma: A report from the Childhood Cancer Survivor Study. Cancer 2017, 123, 2551–2560. [Google Scholar] [CrossRef]
- Friedman, D.N.; Chastain, K.; Chou, J.F.; Moskowitz, C.S.; Adsuar, R.; Wexler, L.H.; Chou, A.J.; DeRosa, A.; Candela, J.; Magnan, H.; et al. Morbidity and mortality after treatment of Ewing sarcoma: A single-institution experience. Pediatric Blood Cancer 2017, 64, e26562. [Google Scholar] [CrossRef]
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.J.; Lessnick, S.L. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2013, 32, 5089–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A first-generation pediatric cancer dependency map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef]
- Grant, C.V.; Cai, S.; Risinger, A.L.; Liang, H.; O’Keefe, B.R.; Doench, J.G.; Cichewicz, R.H.; Mooberry, S.L. CRISPR-Cas9 Genome-Wide Knockout Screen Identifies Mechanism of Selective Activity of Dehydrofalcarinol in Mesenchymal Stem-like Triple-Negative Breast Cancer Cells. J. Nat. Prod. 2020, 83, 3080–3092. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Type | GI50 (nM) | TGI (nM) | LC50 (nM) |
|---|---|---|---|---|
| RD-ES | ES | 7.8 | 32 | 380 |
| SK-ES-1 | ES | 7.6 | 20 | 58 |
| A-673 | ES | 11 | 37 | 150 |
| TC32 | ES | 4.0 | 12 | 48 |
| TC32-NR0B1 | ES | 10 | 23 | 57 |
| TC32-CMV | ES | 5.0 | 10.0 | 20 |
| SJCRH30 | RMS | 120 | 220 | 430 |
| D283 | Med | 100 | 260 | 880 |
| SK-N-BE(2)-C | NB | 270 | 570 | 1200 |
| Hep293TT | HB | 240 | 540 | 1300 |
| SK-OV-3 | OV | 250 | 550 | 1300 |
| SK-OV-3-MDR1-6/6 | OV | 320 | 770 | 1900 |
| MDA-MB-453 | BR | 230 | 790 | 2700 |
| MDA-MB-231 | BR | 400 | 1200 | 3700 |
| HCC1806 | BR | 80 | 220 | 650 |
| BT-549 | BR | 100 | 380 | 1500 |
| HCC1937 | BR | 200 | 680 | 2400 |
| CAL-51 | BR | 230 | 810 | 2800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robles, A.J.; Dai, W.; Haldar, S.; Ma, H.; Anderson, V.M.; Overacker, R.D.; Risinger, A.L.; Loesgen, S.; Houghton, P.J.; Cichewicz, R.H.; et al. Altertoxin II, a Highly Effective and Specific Compound against Ewing Sarcoma. Cancers 2021, 13, 6176. https://doi.org/10.3390/cancers13246176
Robles AJ, Dai W, Haldar S, Ma H, Anderson VM, Overacker RD, Risinger AL, Loesgen S, Houghton PJ, Cichewicz RH, et al. Altertoxin II, a Highly Effective and Specific Compound against Ewing Sarcoma. Cancers. 2021; 13(24):6176. https://doi.org/10.3390/cancers13246176
Chicago/Turabian StyleRobles, Andrew J., Wentao Dai, Saikat Haldar, Hongyan Ma, Victoria M. Anderson, Ross D. Overacker, April L. Risinger, Sandra Loesgen, Peter J. Houghton, Robert H. Cichewicz, and et al. 2021. "Altertoxin II, a Highly Effective and Specific Compound against Ewing Sarcoma" Cancers 13, no. 24: 6176. https://doi.org/10.3390/cancers13246176