
The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Processing
2.3. Proteomic Analysis
2.4. Pathway Analysis
2.5. Kidney Tumor ECM Analysis
2.6. Flow Cytometry
2.7. Primary Cell Culture
2.8. Cell Line Culture
2.9. Immunocytochemistry Validation of Primary Cell Lines
2.10. Cell Line Attachment Assays
2.11. Primary Cell ECM Attachment and Growth
2.12. D Cultures
2.13. Live/Dead and Structure Analysis
2.14. D Dome Immunostaining
2.15. EdU Incorporation and Detection
2.16. scRNA-seq Analysis
2.17. ProteinAtlas Analysis
2.18. Immunohistochemistry
3. Results
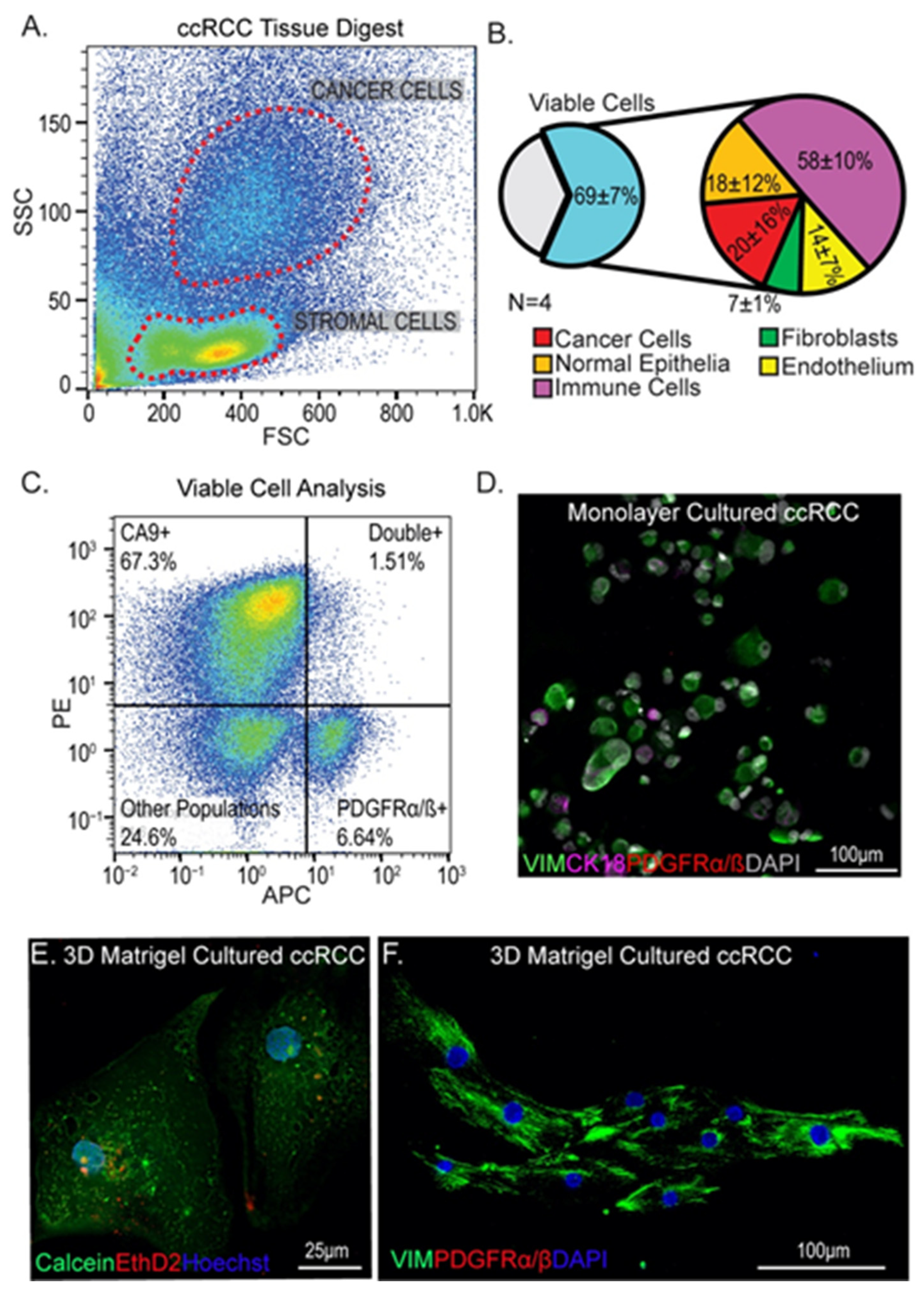
3.1. Isolation and Culture of Cells from ccRCC Tumors Using Standard Methods
3.2. Differences in ECM Composition between ccRCC and Neighboring Healthy Cortex
3.3. Transcriptional Analysis Indicates That Tumor Fibroblasts Are Major Contributors to ccRCC ECM
3.4. Binding of Tumor and Stromal Cells to ccRCC ECM Components
3.5. ccRCC ECM Blend Binds Diverse Cell Types Isolated from Patient Tumors
3.6. Design of a 3D Model of ccRCC with Native ECM Environment
3.7. D ccRCC ECM Model Maintains ccRCC Fibroblast Representation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Haake, S.M.; Rathmell, W.K. Renal Cancer Subtypes: Should We be Lumping or Splitting for Therapeutic Decision Making? Cancer 2017, 123, 200–209. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved Identification of Von Hippel-Lindau Gene Alterations in Clear Cell Renal Tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G., Jr.; Goldberg, M.A. Negative Regulation of Hypoxia-Inducible Genes by the Von Hippel-Lindau Protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF Axis in Renal Cell Carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Lindgren, D.; Sjölund, J.; Axelson, H. Tracing Renal Cell Carcinomas back to the Nephron. Trends Cancer 2018, 4, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Delahunt, B.; Srigley, J.R. The Evolving Classification of Renal Cell Neoplasia. Semin. Diagn. Pathol. 2015, 32, 90–102. [Google Scholar] [CrossRef]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the Malignant Phenotype of Human Breast Cells in Three-Dimensional Culture and in Vivo by Integrin Blocking Antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettouchi, A.; Klein, S.; Guo, W.; Lopez-Lago, M.; Lemichez, E.; Westwick, J.K.; Giancotti, F.G. Integrin-Specific Activation of Rac Controls Progression through the G(1) Phase of the Cell Cycle. Mol. Cell 2001, 8, 115–127. [Google Scholar] [CrossRef]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of Vascular Integrin Alpha V Beta 3 for Angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin Alpha V Beta 3 Antagonists Promote Tumor Regression by Inducing Apoptosis of Angiogenic Blood Vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- Wang, Y.; McNiven, M.A. Invasive Matrix Degradation at Focal Adhesions Occurs via Protease Recruitment by a FAK-p130Cas Complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional Homeostasis and the Malignant Phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 Integrin Amplifies ErbB2 Signaling to Promote Mammary Tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The Extracellular Matrix: Tools and Insights for the “Omics” Era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic Analyses of ECM during Pancreatic Ductal Adenocarcinoma Progression Reveal Different Contributions by Tumor and Stromal Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular Matrix Directs Phenotypic Heterogeneity of Activated Fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of Patient-Derived Cancer Organoids for Drug-Screening Applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Orkin, R.W.; Gehron, P.; McGoodwin, E.B.; Martin, G.R.; Valentine, T.; Swarm, R. A Murine Tumor Producing a Matrix of Basement Membrane. J. Exp. Med. 1977, 145, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Xia, X.; Li, F.; He, J.; Aji, R.; Gao, D. Organoid Technology in Cancer Precision Medicine. Cancer Lett. 2019, 457, 20–27. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostama, B.; Beauchemin, M.; Bouchard, C.; Bernier, E.; Vary, C.P.H.; May, M.; Houseknecht, K.L. Understanding Mechanisms Underlying Non-Alcoholic Fatty Liver Disease (NAFLD) in Mental Illness: Risperidone and Olanzapine Alter the Hepatic Proteomic Signature in Mice. Int. J. Mol. Sci. 2020, 21, 9362. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Turner, J.E.; Harrington, A.; Davis-Knowlton, J.; Lindner, V.; Gridley, T.; Vary, C.P.H.; Liaw, L. Notch2 and Proteomic Signatures in Mouse Neointimal Lesion Formation. Arter. Thromb. Vasc. Biol. 2018, 38, 1576–1593. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Burton, L.; Lau, A.; Tate, S. SWATH-ID: An Instrument Method Which Combines Identification and Quantification in a Single Analysis. Proteomics 2017, 10, e150052. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, M.; Geguchadze, R.; Guntur, A.R.; Nevola, K.; Le, P.T.; Barlow, D.; Rue, M.; Vary, C.P.H.; Lary, C.W.; Motyl, K.J.; et al. Exploring Mechanisms of Increased Cardiovascular Disease Risk with Antipsychotic Medications: Risperidone Alters the Cardiac Proteomic Signature in Mice. Pharmacol. Res. 2020, 152, 104589. [Google Scholar] [CrossRef]
- Ivosev, G.; Burton, L.; Bonner, R. Dimensionality Reduction and Visualization in Principal Component Analysis. Anal. Chem. 2008, 80, 4933–4944. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.T.; Naba, A. MatrisomeDB: The ECM-Protein Knowledge Database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the Matrisome—an Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.D.; Elliott, A.Y.; Stein, N.; Fraley, E.E. In Vitro Cultivation of Human Renal Cell Cancer. II. Characterization of cell lines. In Vitro 1978, 14, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, Y.; Zhang, H.; Zhang, Y.; Xu, P.; Chen, J.; Poh, Y.C.; Tang, K.; Wang, N.; Huang, B. Soft Fibrin Gels Promote Selection and Growth of Tumorigenic Cells. Nat. Mater. 2012, 11, 734–741. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-Cell Transcriptomes from Human Kidneys Reveal the Cellular Identity of Renal Tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Fetting, J.L.; Guay, J.A.; Karolak, M.J.; Iozzo, R.V.; Adams, D.C.; Maridas, D.E.; Brown, A.C.; Oxburgh, L. FOXD1 Promotes Nephron Progenitor Differentiation by Repressing Decorin in the Embryonic Kidney. Development 2014, 141, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeehan, W.L.; Ham, R.G. Methods for Reducing the Serum Requirement for Growth in Vitro of Nontransformed Diploid Fibroblasts. Dev. Biol. Stand. 1976, 37, 97–98. [Google Scholar]
- Whateley, J.G.; Knox, P. Isolation of a Serum Component that Stimulates the Spreading of Cells in Culture. Biochem. J. 1980, 185, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.X.; Song, X.; Pascovici, D.; Zaw, T.; Care, N.; Krisp, C.; Molloy, M.P. SWATH Mass Spectrometry Performance Using Extended Peptide MS/MS Assay Libraries. Mol. Cell Proteom. 2016, 15, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Krasny, L.; Bland, P.; Kogata, N.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. SWATH Mass Spectrometry as a Tool for Quantitative Profiling of the Matrisome. J. Proteom. 2018, 189, 11–22. [Google Scholar] [CrossRef]
- Courtney, K.D.; Bezwada, D.; Mashimo, T.; Pichumani, K.; Vemireddy, V.; Funk, A.M.; Wimberly, J.; McNeil, S.S.; Kapur, P.; Lotan, Y.; et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab. 2018, 28, 793–800.e2. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The Matrisome: In Silico Definition and in Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Cornil, I.; Theodorescu, D.; Man, S.; Herlyn, M.; Jambrosic, J.; Kerbel, R.S. Fibroblast Cell Interactions with Human Melanoma Cells Affect Tumor Cell Growth as a Function of Tumor Progression. Proc. Natl. Acad. Sci. USA 1991, 88, 6028–6032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolomé, R.A.; Peña, C.; Fernandez-Aceñero, M.J.; Padilla, G.; Peláez-García, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R.; et al. LOXL2 Is Highly Expressed in Cancer-Associated Fibroblasts and Associates to Poor Colon Cancer Survival. Clin. Cancer Res. 2015, 21, 4892–4902. [Google Scholar] [CrossRef] [Green Version]
- Rupp, C.; Scherzer, M.; Rudisch, A.; Unger, C.; Haslinger, C.; Schweifer, N.; Artaker, M.; Nivarthi, H.; Moriggl, R.; Hengstschläger, M.; et al. IGFBP7, A Novel Tumor Stroma Marker, with Growth-Promoting Effects in Colon Cancer Through a Paracrine Tumor-Stroma Interaction. Oncogene 2015, 34, 815–825. [Google Scholar] [CrossRef]
- Lindop, G.B.; Fleming, S. Renin in Renal Cell Carcinoma—An Immunocytochemical Study Using an Antibody to Pure Human renin. J. Clin. Pathol. 1984, 37, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Raghavan, S.; Snyder, C.S.; Mehta, G. Integrated Cancer Tissue Engineering Models for Precision Medicine. PLoS ONE 2019, 14, e0216564. [Google Scholar]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient Derived Organoids to Model Rare Prostate Cancer Phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-Derived Lung Cancer Organoids as in Vitro Cancer Models for Therapeutic Screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef]
- Janmey, P.A.; Winer, J.P.; Weisel, J.W. Fibrin Gels and Their Clinical and Bioengineering Applications. J. R. Soc. Interface 2009, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Gimona, M.; Buccione, R.; Courtneidge, S.A.; Linder, S. Assembly and Biological Role of Podosomes and Invadopodia. Curr. Opin. Cell Biol. 2008, 20, 235–241. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.R.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor Morphology and Phenotypic Evolution Driven by Selective Pressure from the Microenvironment. Cell 2006, 127, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Miki, J.; Furusato, B.; Li, H.; Gu, Y.; Takahashi, H.; Egawa, S.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Rhim, J.S. Identification of Putative Stem Cell Markers, CD133 and CXCR4, in hTERT-Immortalized Primary Nonmalignant and Malignant Tumor-Derived Human Prostate Epithelial Cell Lines and in Prostate Cancer Specimens. Cancer Res. 2007, 67, 3153–3161. [Google Scholar] [CrossRef] [Green Version]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in Renal Cancer Stem Cells and the Implications for Human Patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Kaelin, W.G., Jr. The VHL/HIF Axis in Clear Cell Renal Carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad Targeting of Angiogenesis for Cancer Prevention and Therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Battle, D.; Figlin, R.A.; George, D.J.; Hammers, H.; Hutson, T.; Jonasch, E.; Joseph, R.W.; McDermott, D.F.; Motzer, R.J.; et al. The Society for Immunotherapy of Cancer Consensus Statement on Immunotherapy for the Treatment of Advanced Renal Cell Carcinoma (RCC). J. Immunother. Cancer 2019, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental Regulation of Tumour Angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The Polarization of Immune Cells in the Tumour Environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Esser, L.K.; Branchi, V.; Leonardelli, S.; Pelusi, N.; Simon, A.G.; Klümper, N.; Ellinger, J.; Hauser, S.; Gonzalez-Carmona, M.A.; Ritter, M.; et al. Cultivation of Clear Cell Renal Cell Carcinoma Patient-Derived Organoids in an Air-Liquid Interface System as a Tool for Studying Individualized Therapy. Front. Oncol. 2020, 10, 1775. [Google Scholar] [CrossRef]
- Batchelder, C.A.; Martinez, M.L.; Duru, N.; Meyers, F.J.; Tarantal, A.F. Three Dimensional Culture of Human Renal Cell Carcinoma Organoids. PLoS ONE 2015, 10, e0136758. [Google Scholar] [CrossRef]
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix Metalloproteinase-11/Stromelysin-3 Exhibits Collagenolytic Function against Collagen VI under Normal and Malignant Conditions. Oncogene 2008, 27, 6347–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. TEM8 Interacts with the Cleaved C5 Domain of Collagen Alpha 3(VI). Cancer Res. 2004, 64, 817–820. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-Derived Collagen VI Affects Early Mammary Tumor Progression in Vivo, Demonstrating a Critical Interaction in the Tumor/Stroma Microenvironment. J. Clin. Investig. 2005, 115, 1163–1176. [Google Scholar] [CrossRef] [Green Version]
- Rühl, M.; Sahin, E.; Johannsen, M.; Somasundaram, R.; Manski, D.; Riecken, E.O.; Schuppan, D. Soluble Collagen VI Drives Serum-Starved Fibroblasts through S Phase and Prevents Apoptosis via Down-Regulation of Bax. J. Biol. Chem. 1999, 274, 34361–34368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, W.K.; Bonaldo, P.; Stallcup, W.B. Collagen VI Ablation Retards Brain Tumor Progression due to Deficits in Assembly of the Vascular Basal Lamina. Am. J. Pathol. 2012, 180, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Wang, H.; Shen, Y.; Zhang, H.; Shi, G.; Zhu, Y.; Dai, B.; Ye, D. Upregulation of COL6A1 Is Predictive of Poor Prognosis in Clear Cell Renal Cell Carcinoma Patients. Oncotarget 2015, 6, 27378–27387. [Google Scholar] [CrossRef] [Green Version]
- Han, S.W.; Roman, J. Fibronectin Induces Cell Proliferation and Inhibits Apoptosis in Human Bronchial Epithelial Cells: Pro-oncogenic Effects Mediated by PI3-Kinase and NF-Kappa B. Oncogene 2006, 25, 4341–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Khuri, F.R.; Roman, J. Fibronectin Stimulates Non-Small Cell Lung Carcinoma Cell Growth through Activation of Akt/Mammalian Target of Rapamycin/S6 Kinase and Inactivation of LKB1/AMP-Activated Protein Kinase Signal Pathways. Cancer Res. 2006, 66, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Chiquet-Ehrismann, R.; Moyano, J.V.; Garcia-Pardo, A.; Orend, G. Interference of Tenascin-C with Syndecan-4 Binding to Fibronectin Blocks Cell Adhesion and Stimulates Tumor Cell Proliferation. Cancer Res. 2001, 61, 8586–8594. [Google Scholar]
- Yoshida, T.; Yoshimura, E.; Numata, H.; Sakakura, Y.; Sakakura, T. Involvement of Tenascin-C in Proliferation and Migration of Laryngeal Carcinoma Cells. Virchows Arch. 1999, 435, 496–500. [Google Scholar] [CrossRef]
- Guo, S.K.; Shen, M.F.; Yao, H.W.; Liu, Y.S. Enhanced Expression of TGFBI Promotes the Proliferation and Migration of Glioma Cells. Cell Physiol. Biochem. 2018, 49, 1097–1109. [Google Scholar] [CrossRef]
- Bao, S.; Ouyang, G.; Bai, X.; Huang, Z.; Ma, C.; Liu, M.; Shao, R.; Anderson, R.M.; Rich, J.N.; Wang, X.F. Periostin Potently Promotes Metastatic Growth of Colon Cancer by Augmenting Cell Survival via the Akt/PKB Pathway. Cancer Cell 2004, 5, 329–339. [Google Scholar] [CrossRef]
- Jia, Y.Y.; Yu, Y.; Li, H.J. POSTN Promotes Proliferation and Epithelial-Mesenchymal Transition in Renal Cell Carcinoma through ILK/AKT/mTOR Pathway. J. Cancer 2021, 12, 4183–4195. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, A.; Bond, K.; Chiba, T.; Sims-Lucas, S.; Oxburgh, L.; Coburn, J.M. Development of a Mechanically Matched Silk Scaffolded 3D Clear Cell Renal Cell Carcinoma Model. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 126, 112141. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal Myofibroblasts Are Drivers of Invasive Cancer Growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef]
- Siljee, S.; Milne, B.; Brasch, H.D.; Bockett, N.; Patel, J.; Davis, P.F.; Kennedy-Smith, A.; Itinteang, T.; Tan, S.T. Expression of Components of the Renin-Angiotensin System by Cancer Stem Cells in Renal Clear Cell Carcinoma. Biomolecules 2021, 11, 537. [Google Scholar] [CrossRef]
- Everett, A.D.; Carey, R.M.; Chevalier, R.L.; Peach, M.J.; Gomez, R.A. Renin Release and Gene Expression in Intact Rat Kidney Microvessels and Single Cells. J. Clin. Investig. 1990, 86, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Sequeira López, M.L.; Pentz, E.S.; Nomasa, T.; Smithies, O.; Gomez, R.A. Renin Cells Are Precursors for Multiple Cell Types That Switch to the Renin Phenotype When Homeostasis Is Threatened. Dev. Cell 2004, 6, 719–728. [Google Scholar] [CrossRef]
- McKay, R.R.; Rodriguez, G.E.; Lin, X.; Kaymakcalan, M.D.; Hamnvik, O.P.; Sabbisetti, V.S.; Bhatt, R.S.; Simantov, R.; Choueiri, T.K. Angiotensin System Inhibitors and Survival Outcomes in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Khanna, P.; Soh, H.J.; Chen, C.H.; Saxena, R.; Amin, S.; Naughton, M.; Joslin, P.N.; Moore, A.; Bakouny, Z.; O’Callaghan, C.; et al. ACE2 Abrogates Tumor Resistance to VEGFR Inhibitors Suggesting Angiotensin-(1–7) as a Therapy for Clear Cell Renal Cell Carcinoma. Sci. Transl. Med. 2021, 13, eabc0170. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Diagnosis | Stage | Grade | Sex | Race | Age | Applications |
|---|---|---|---|---|---|---|---|
| Tp17-S560 | ccRCC | pT3 Nx Mx | 3 | M | White | 72 | Mass Spec |
| Tp18-S435 | ccRCC | pT3a NX | 4 | M | White | 50–59 | Mass Spec |
| R19-6098 | ccRCC | pT3 pN0 | 2 | N/A | White | 29 | Mass Spec |
| Tp19-S80 | ccRCC | pT3a N0 Mx | 4 | M | White | 69 | Mass Spec |
| Tp19-S79 | ccRCC | pT3a Nx | 4 | F | White | 68 | Mass Spec |
| Tp18-S109 | ccRCC | pT3a | 2 | M | White | 60–69 | Mas Spec, Primary Culture |
| Tp18-S114 | ccRCC | pT3a | 3 | M | White | 80–89 | Mass Spec, Primary Culture |
| Tp18-S108 | ccRCC | pT1a Nx Mx | 2 | F | White | 60–69 | Primary Culture |
| R18-6256 | ccRCC | pT1a pNC | 1 | N/A | N/A | 61 | Flow Cytometry |
| R19-6200 | ccRCC | pT1b Nx | 2 | M | White | 61 | Flow Cytometry |
| Tp18-S601 | ccRCC | pT1b Nx | 2 | M | White | 61 | Flow Cytometry |
| R18-1453 | ccRCC | pT2a pNx | 3 | F | White | 83 | Flow Cytometry |
| Tp21-S145 | ccRCC | pT3a Nx Mx | 2 | F | White | 76 | 3D Culture |
| Tp20-S199 | ccRCC | pT3a Nx | 2 | M | N/A | 74 | 3D Culture |
| Tp21-S27 | ccRCC (Syndromic VHL) | pT1a Nx Mx | 4 | M | White | 68 | 3D Culture |
| Tp20-S215 | ccRCC | pT4 Nx M1 | 4 | M | White | 52 | 3D Culture |
| Tp20-S238 | ccRCC | PT3 + Nx M1 | 4 | M | White | 71 | 3D Culture |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bond, K.H.; Chiba, T.; Wynne, K.P.H.; Vary, C.P.H.; Sims-Lucas, S.; Coburn, J.M.; Oxburgh, L. The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth. Cancers 2021, 13, 5873. https://doi.org/10.3390/cancers13235873
Bond KH, Chiba T, Wynne KPH, Vary CPH, Sims-Lucas S, Coburn JM, Oxburgh L. The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth. Cancers. 2021; 13(23):5873. https://doi.org/10.3390/cancers13235873
Chicago/Turabian StyleBond, Kyle H., Takuto Chiba, Kieran P. H. Wynne, Calvin P. H. Vary, Sunder Sims-Lucas, Jeannine M. Coburn, and Leif Oxburgh. 2021. "The Extracellular Matrix Environment of Clear Cell Renal Cell Carcinoma Determines Cancer Associated Fibroblast Growth" Cancers 13, no. 23: 5873. https://doi.org/10.3390/cancers13235873