
Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets


 ,
,
Abstract
:Simple Summary
Abstract
1. Cancer-Associated Fibroblasts: Biology of the Fibrotic Tumor Stroma
2. CAF Functional Complexity
3. Heterotypic Spheroids: A Specialized CAF–Tumor Microenvironment
4. CAFs Confer Resistance to Chemotherapy
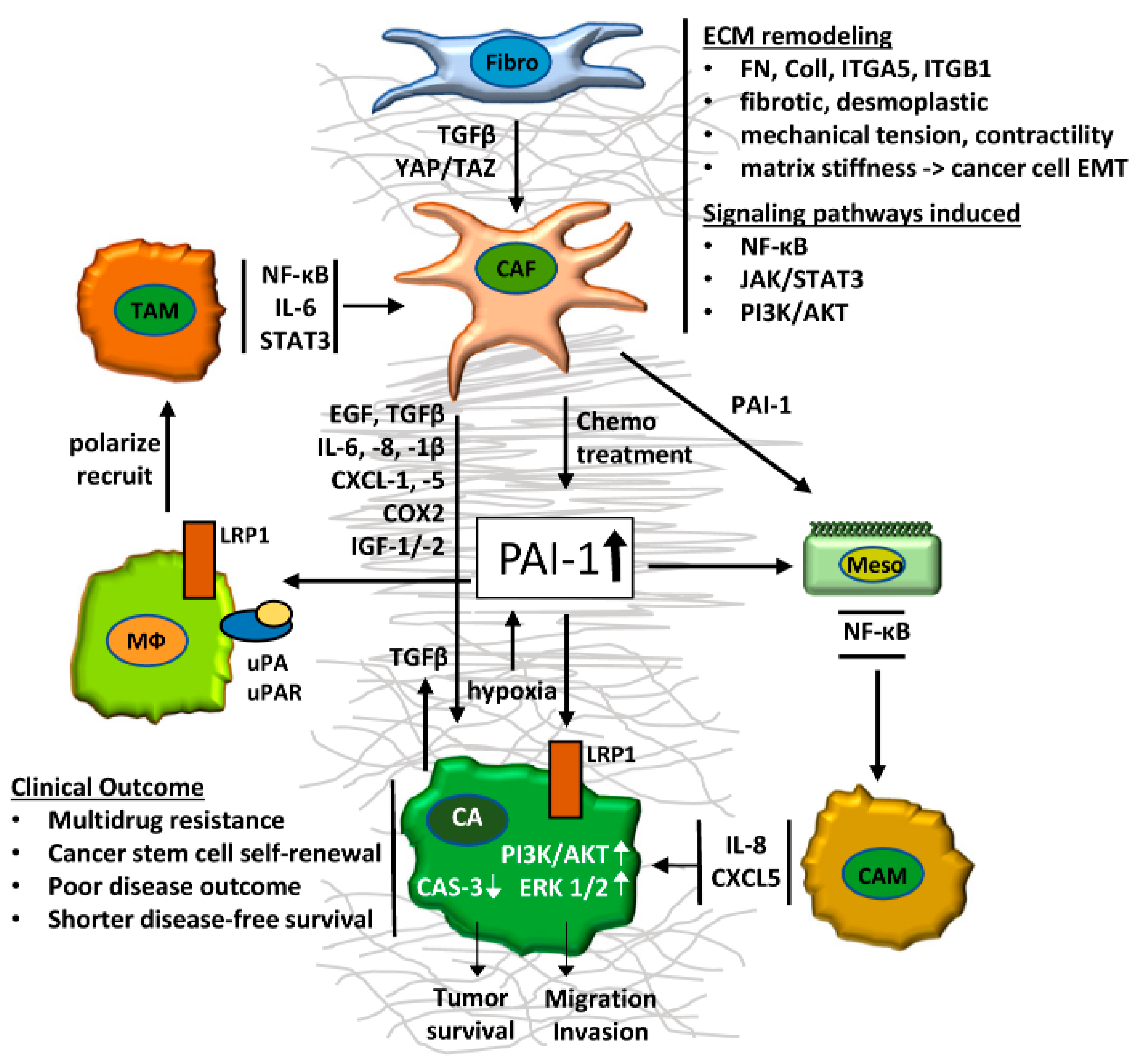
5. CAF-Derived PAI-1 as a Poor Prognosis Biomarker
6. The CAF/PAI-1 Axis in the Tumor Immune Response
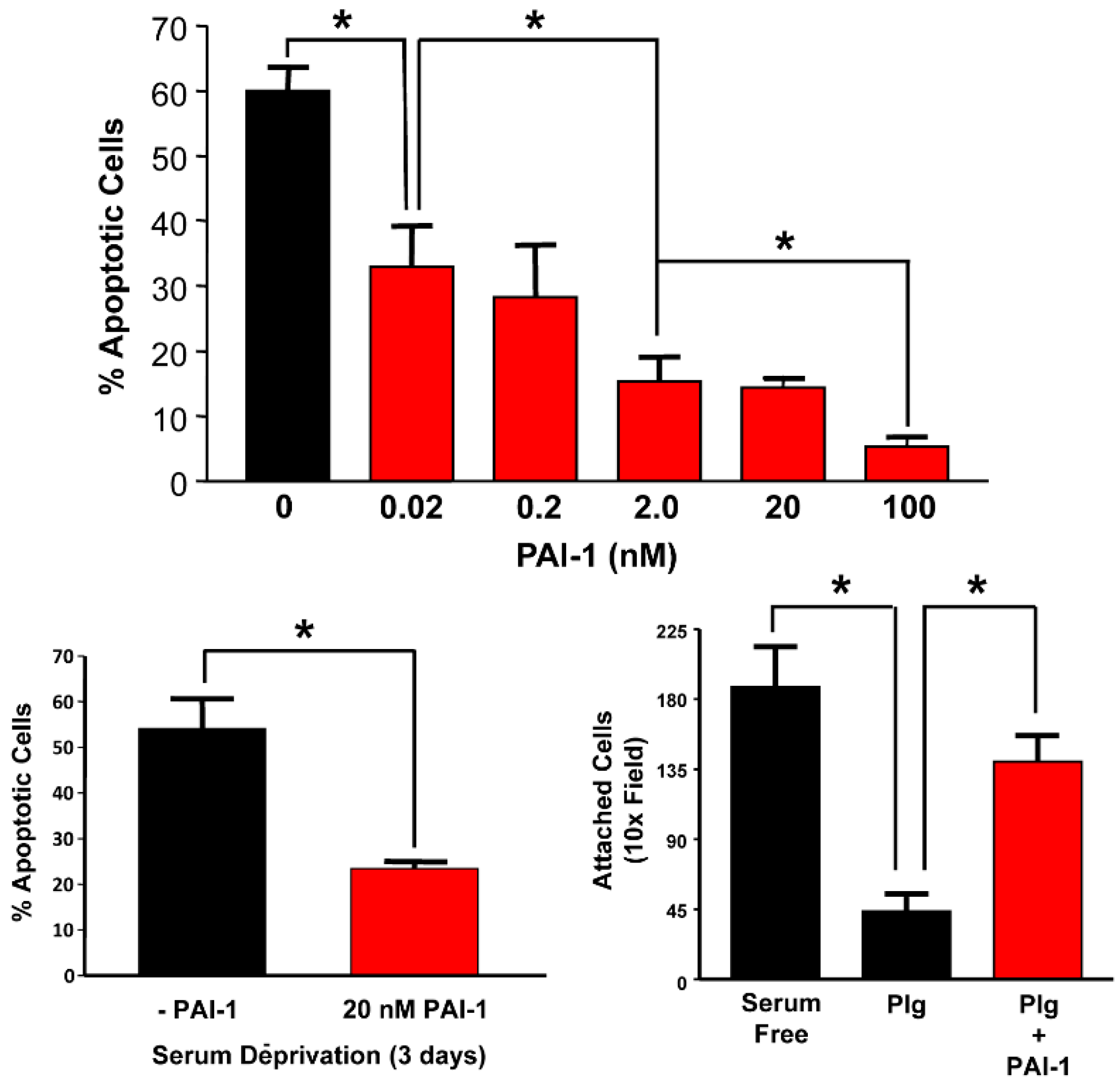
7. Role of PAI-1 and the CAF/PAI-1 Axis in Tumor Survival and Cell Migration
8. Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2018, 18, 99–115. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 21, 1503–1523. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Denton, A.C.; Roberts, E.W.; Fearon, D.T. Stromal cells in the tumor microenvironment. Adv. Exp. Med. Biol. 2018, 1060, 99–114. [Google Scholar]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 186–196. [Google Scholar] [CrossRef]
- Linares, J.; Marín-Jiménez, J.A.; Badia-Ramentol, J.; Calon, A. Determinants and functions of CAFs secretome during cancer progression and therapy. Front. Cell Dev. Biol. 2021, 8, 621070. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar] [PubMed]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J., 3rd; et al. Cancer-associated fibroblasts: Versatile players in the tumor microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Bochim. Biophys. Acta Rev. Cancer 2020, 1872, 188356. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts-heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Pradhan, R.N.; Krishnamurty, A.T.; Fletcher, A.L.; Turley, S.J.; Müller, S. A bird’s eye view of fibroblast heterogeneity: A pan-disease, pan-cancer perspective. Immunol. Rev. 2021, 302, 299–320. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X. Transforming growth factor-β signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
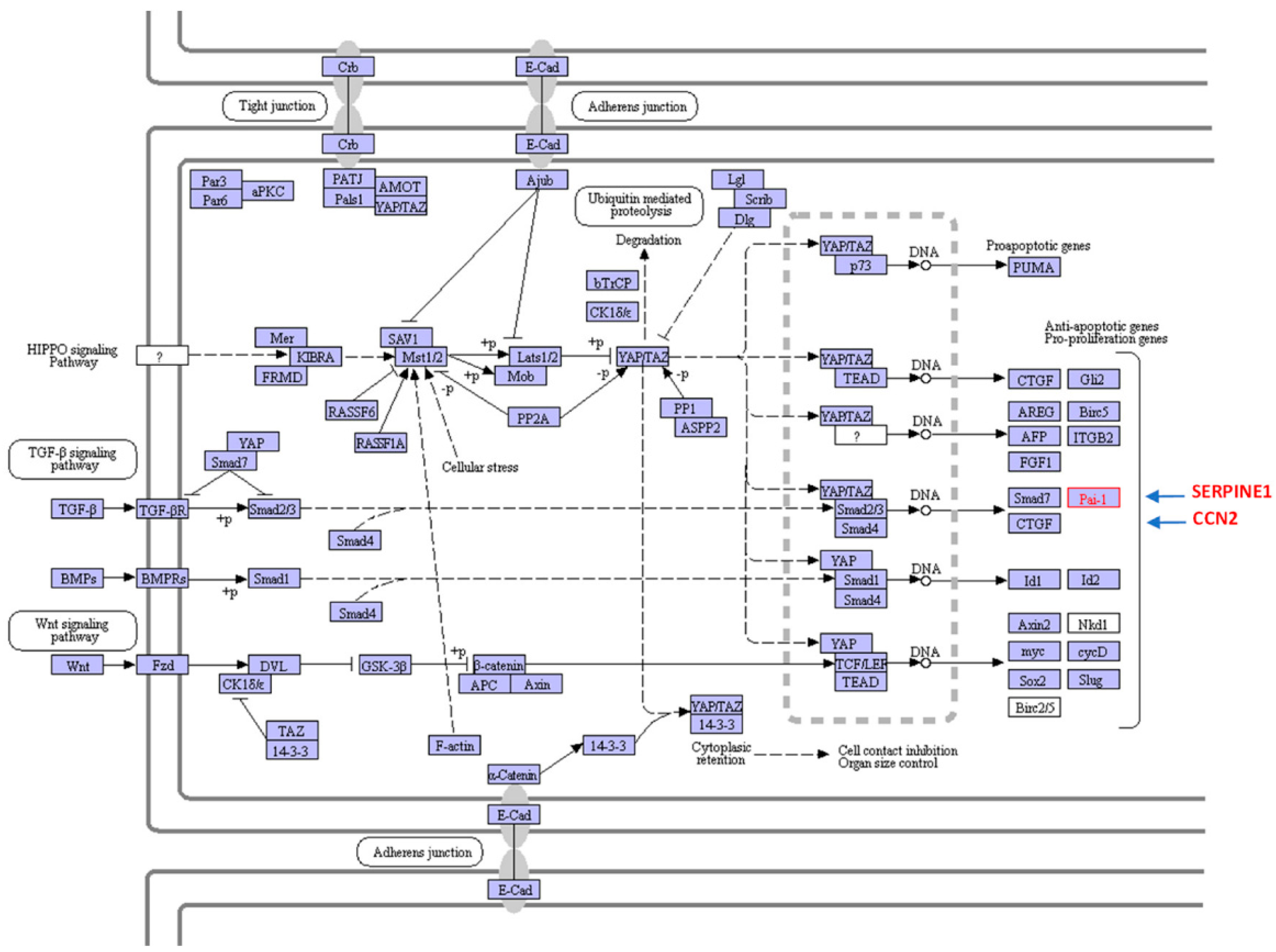
- Noguchi, S.; Saito, A.; Nagase, T. Yap/Taz signaling as a molecular link between fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in fibrosis: TGF-β, Wnt, and Yap/Taz converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Mallikarjuna, P.; Raviprakash, T.S.; Aripaka, K.; Ljungberg, B.; Landstrom, M. Interactions between TGF-β type I receptor and hypoxia-inducible factor-α mediates a synergistic crosstalk leading to poor prognosis for patients with clear cell renal cell carcinoma. Cell Cycle 2019, 18, 2141–2156. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. Yap and Taz: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Gifford, C.C.; Lian, F.; Tang, J.; Costello, A.; Goldschmeding, R.; Samarakoon, R.; Higgins, P.J. PAI-1 induction during kidney injury promotes fibrotic epithelial dysfunction via deregulation of klotho, p53, and TGF-β-receptor signaling. FASEB J. 2021, 35, e21725. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.E.; Tang, J.; Mian, B.M.; Higgins, S.P.; Gifford, C.C.; Conti, D.J.; Meldrum, K.K.; Samarakoon, R.; Higgins, P.J. TGF-β1-p53 cooperativity regulates a profibrotic genomic program in the kidney: Molecular mechanisms and clinical implications. FASEB J. 2019, 33, 10596–10606. [Google Scholar] [CrossRef] [Green Version]
- Anorga, S.; Overstreet, J.M.; Falke, L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-Taz pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lagares, D.; Cho, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through Yap and Taz drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef] [Green Version]
- Samarakoon, R.; Helo, S.; Dobberfuhl, A.D.; Khakoo, N.S.; Falke, L.; Overstreet, J.M.; Goldschmeding, R.; Higgins, P.J. Loss of tumour suppressor PTEN expression in renal injury initiates SMAD3- and p53-dependent fibrotic responses. J. Pathol. 2015, 236, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.-J.; Kwon, E.-J.; Kwon, O.-K.; Lee, H.; Choi, J.-Y.; Kim, Y.-J.; Kim, W.; Cha, H.-J. Crosstalk between Yap and TGFβ regulates SERPINE1 expresssion in mesenchymal lung cancer cells. Int. J. Oncol. 2021, 58, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.E.; Tang, J.; Higgins, S.P.; Gifford, C.C.; Mian, B.M.; Jones, D.M.; Zhang, W.; Costello, A.; Conti, D.J.; Samarakoon, R.; et al. The genomic response to TGF-β1 dictates failed repair and progression of fibrotic disease in the obstructed kidney. Front. Cell Dev. Biol. 2021, 9, 678524. [Google Scholar] [CrossRef]
- Marquard, S.; Thomann, S.; Weiler, S.M.E.; Bissinger, M.; Lutz, T.; Sticht, C.; Tóth, M.; de la Torre, C.; Gretz, N.; Straub, B.K.; et al. Yes-associated protein (YAP) induces a secretome phenotype and transcriptionally regulates plasminogen activator inhibitor-1 (PAI-1) expression in hepatocarcinogenesis. Cell Commun. Signal. 2020, 18, 166. [Google Scholar] [CrossRef]
- Tadeo, I.; Berbegall, A.P.; Escudero, L.M.; Alvaro, T.; Noguera, R. Biotensegrity of the extracellular matrix: Physiology, dynamic mechanical balance, and implications in oncology and mechanotherapy. Front. Oncol. 2014, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020, 39, 112. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Philippeos, C.; Telerman, S.B.; Oulès, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Investig. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Tabib, T.; Morse, C.; Wang, T.; Chen, W.; Lafyatis, R. SFRP2/DPP4 and FMOL/LSP1 define major fibroblast populations in human skin. J. Investig. Dermatol. 2018, 138, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Peyser, R.; MacDonnell, S.; Gao, Y.; Cheng, L.; Kim, Y.; Kaplan, T.; Ruan, Q.; Wei, Y.; Ni, M.; Adler, C.; et al. Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am. J. Respir. Cell. Mol. Biol. 2019, 61, 74–85. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The role of tumor microenvironment in cancer metastasis: Molecular mechanisms and therapeutic opportunities. Cancers 2021, 13, 2053. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Da Cunha, B.R.; Domingos, C.; Stefanini, A.C.B.; Henrique, T.; Polachini, G.M.; Castelo-Branco, P.; Tajara, E.H. Cellular interactions in the tumor microenvironment: The role of secretome. J. Cancer 2019, 10, 4574–4587. [Google Scholar] [CrossRef] [Green Version]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C.A.-O. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, H.; Zhu, L.; Li, J.; Ma, S. Cancer-associated fibroblasts in non-small cell lung cancer: Recent advances and future perspectives. Cancer Lett. 2021, 514, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Sun, J.; Xie, M.; Yu, S.; Tang, Q.; Chen, L. Plau promotes cell proliferation and epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Front. Genet. 2021, 12, 651882. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, D.E.; Brown, N.J. Effects of acute angiotensin II type 1 receptor antagonism and angiotensin converting enzyme inhibition on plasma fibrinolytic parameters in patients with heart failure. Circulation 2000, 102, E43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caja, L.C.O.; Bertran, E.; Murillo, M.M.; Miró-Obradors, M.J.; Palacios, E.; Fabregat, I. Differential intracellular signalling induced by TGF-β in rat adult hepatocytes and hepatoma cells: Implications in liver carcinogenesis. Cell Signal. 2007, 19, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labibi, B.; Bashkurov, M.; Wrana, J.L.; Attisano, L. Modeling the control of TGF-β/Smad nuclear accumulation by the Hippo pathway effectors, Taz/Yap. iScience 2020, 23, 101416. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Yu, X.; Yang, F.; Zhang, Z.; Shen, J.; Sun, J.; Choksi, S.; Jitkaew, S.; Shu, Y. Reprogramming of normal fibroblasts into cancer-associated fibroblasts by miRNAs-mediated CCL2/VEGFA signaling. PLoS Genet. 2016, 12, e1006244. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Landry, N.M.; Dixon, I.M.C. Fibroblast mechanosensing, Ski and Hippo signaling and the cardiac fibroblast phenotype: Looking beyond TGF-β. Cell Signal. 2020, 76, 109802. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, L.; Xu, R.; Zhang, C.; Xie, Y.; Liu, K.; Li, T.; Hu, W.; Zhen, C.; Wang, F.S. Mesenchymal stem cell therapy for severe Covid-19. Signal. Transduct. Target. Ther. 2021, 6, 339. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Shaked, Y.; Tsai, K.K. Targeting the interplay between cancer fibroblasts, mesenchymal stem cells, and cancer stem cells in desmoplastic cancers. Front. Oncol. 2019, 9, 688. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Mooney, D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. USA 2016, 113, 12126–12131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, O.; Koshy, S.T.; Branco da Cunha, C.; Shin, J.W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Drain, A.P.; Weaver, V.M. The extracellular matrix modulates the metastatic journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Loh, J.J.; Ma, S. The role of cancer-associated fibroblast as a dynamic player in mediating cancer stemness in the tumor microenvironment. Front. Cell Dev. Biol. 2021, 9, 727640. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7 (Suppl. S11), S44–S47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zustiak, S.; Nossal, R.; Sackett, D.L. Multiwell stiffness assay for the study of cell responsiveness to cytotoxic drugs. Biotechnol. Bioeng. 2014, 111, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Weaver, V.M. Mechanics, malignancy, and metastasis: The force journey of a tumor cell. Cancer Metastasis Rev. 2009, 28, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saénz-de-Santa-María, I.; Celada, L.; Chiara, M.D. The leader position of mesenchymal cells expressing N-cadherin in the collective migration of epithelial cancer. Cells 2020, 9, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-induced Jak/Stat signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yan, L.; Xu, Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 2015, 34, 3315–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengyel, E.; Burdette, J.E.; Kenny, H.A.; Matei, D.; Pilrose, J.; Haluska, P.; Nephew, K.P.; Hales, D.B.; Stack, M.S. Epithelial ovarian cancer experimental models. Oncogene 2014, 33, 3619–3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.; Hu, Y.; Long, T.; Lu, X.; Tuo, Y.; Li, Y.; Ke, Z. Tumor-associated macrophages induced spheroid formation by CCL18-ZEB1-m-CSF feed-back loop to promote transcoelomic metastasis of ovarian cancer. J. Immunother. Cancer 2021, 9, e003973. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 2019, 16, 688–703. [Google Scholar] [CrossRef] [Green Version]
- Degryse, B.; Neels, J.G.; Czekay, R.-P.; Aertgeerts, K.; Kamikubo, Y.-I.; Loskutoff, D.J. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J. Biol. Chem. 2004, 279, 22595–22604. [Google Scholar] [CrossRef] [Green Version]
- Czekay, R.-P.; Wilkins-Port, C.E.; Higgins, S.P.; Freytag, J.; Overstreet, J.M.; Klein, R.M.; Higgins, C.E.; Samarakoon, R.; Higgins, P.J. PAI-1: An integrator of cell signaling and migration. Int. J. Cell Biol. 2011, 2011, 562481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, A.S.; Fischer, A.; Miller, D.H.; Vang, S.; MacLaughlan, S.; Wu, H.T.; Yu, J.; Steinhoff, M.; Collins, C.; Smith, P.J.; et al. Expression profiling of primary and metastatic ovarian tumors reveals differences indicative of aggressive disease. PLoS ONE 2014, 9, e94476. [Google Scholar] [CrossRef]
- Wang, W.; Kryczek, I.; Dostál, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.; Mukherjee, A.; Lengyel, E. The tumor microenvironment takes center stage in ovarian cancer metastasis. Trends Cancer 2018, 4, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Galván, S.; Carnero, A. Leveraging genomics, transcriptomics, and epigenomics to understand the biology and chemoresistance of ovarian cancer. Cancers 2021, 13, 4029. [Google Scholar] [CrossRef]
- Khella, C.A.; Mehta, G.A.; Mehta, R.N.; Gatza, M.L. Recent advances in integrative multi-omics research in breast and ovarian cancer. J. Pers. Med. 2021, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef]
- Barbone, D.; Yang, T.M.; Morgan, J.R.; Gaudino, G.; Broaddus, V.C. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008, 283, 13021–13030. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Sproul, D.; Grun, B.; Notaridou, M.; Benjamin, E.; Jacobs, I.J.; Dafou, D.; Sims, A.H.; Gayther, S.A. Modelling genetic and clinical heterogeneity in epithelial ovarian cancers. Carcinogenesis 2011, 32, 1540–1549. [Google Scholar] [CrossRef]
- Matte, I.; Legault, C.M.; Garde-Granger, P.; Laplante, C.; Bessette, P.; Rancourt, C.; Piché, A. Mesothelial cells interact with tumor cells for the formation of ovarian cancer multicellular spheroids in peritoneal effusions. Clin. Exp. Metastasis 2016, 33, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Medina, M.; Yang, J. Mena promotes tumor-intrinsic metastasis through ECM remodeling and haptotaxis. Cancer Discov. 2016, 2016, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Xu, S.; Guo, D.; Zhang, J.; Liu, S. Increased expression of α5β1-integrin is a prognostic marker for patients with gastric cancer. Clin. Transl. Oncol. 2014, 16, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D.; Tolcher, A.W.; Liu, G.; Holen, K.; Schwartz, G.; Albertini, M.; Weiss, G.; Yazji, S.; Ng, C.; Wilding, G. Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: A phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 2008, 14, 7924–7929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoletti, J.P.; Fanaian, N.; Reza, J.; Sause, R.; Almodovar, A.J.; Srivastava, M.; Patel, S.; Veldhuis, P.P.; Griffith, E.; Shao, Y.P.; et al. Pancreatic and bile duct cancer circulating tumor cells (CTC) form immune-resistant multi-cell type clusters in the portal venous circulation. Cancer Biol. Ther. 2018, 19, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Zdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [Green Version]
- Farmer, P.; Yu, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Joyce, M.H.; Lu, C.; James, E.R.; Hegab, R.; Allen, S.C.; Suggs, L.J.; Brock, A. Phenotypic basis for matrix stiffness-dependent chemoresistance of breast cancer cells to doxorubicin. Front. Oncol. 2018, 8, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, Y.; Bonneau, C.; Popova, T.; Rouzier, R.; Stern, M.H.; Mechta-Grigoriou, F. Clinical interest of combining transcriptomic and genomic signatures in high-grade serous ovarian cancer. Front. Genet. 2020, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, K.H.; Chiba, T.; Wynne, K.P.H.; Vary, C.P.H.; Sims-Lucas, S.; Coburn, J.M.; Oxburgh, L.A.-O. The extracellular matrix environment of clear cell renal cell carcinoma determines cancer associated fibroblast growth. Cancers 2021, 13, 5873. [Google Scholar] [CrossRef]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.-H. Tumor-stroma IL-1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemo-resistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amornsupak, K.; Insawang, T.; Thuwajit, P.; O-Charoenrat, P.; Eccles, S.A.; Thuwajit, C. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer 2014, 14, 955. [Google Scholar] [CrossRef]
- Müerköster, S.S.; Werbing, V.; Koch, D.; Sipos, B.; Ammerpohl, O.; Kalthoff, H.; Tsao, M.-S.; Fölsch, U.R.; Schäfer, H. Role of myofibroblasts in innate chemoresistance of pancreatic carcinoma—Epigenetic downregulation of caspases. Int. J. Cancer 2008, 123, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Yong, X.; Wang, P.; Jiang, T.; Yu, W.; Shang, Y.; Han, Y.; Zhang, P.; Li, Q. Fibroblasts weaken the anti-tumor effect of gefitinib on co-cultured non-small cell lung cancer cells. Chin. Med. J. 2014, 127, 2091–2906. [Google Scholar]
- Yan, H.; Guo, B.Y.; Zhang, S. Cancer-associated fibroblasts attenuate cisplatin-induced apoptosis in ovarian cancer cells by promoting stat3 signaling. Biochem. Biophys. Res. Commun. 2016, 470, 947–954. [Google Scholar]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yang, W.-M.; Chen, L.i.-P.; Yang, D.-H.; Zhou, Q.; Zhu, J.; Chen, J.-J.; Huang, R.-C.; Chen, Z.-S.; Huang, R.P. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res. Treat. 2012, 135, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/BCL-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Lin, Q.; Lu, Y.; Li, G.; Huang, I.; Fu, Z.; Chen, R.; Zhou, Q. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-β/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021, 12, 334. [Google Scholar] [CrossRef]
- Du, G.; Cheng, X.; Zhang, Z.; Han, L.; Wu, K.; Li, Y.; Lin, X. TGF-β induced key genes of osteogenic and adipogenic differentiation in human mesenchymal stem cells and miRNA-mRNA regulatory networks. Front. Genet. 2021, 12, 759596. [Google Scholar] [CrossRef] [PubMed]
- Kutz, S.M.; Hordines, J.; McKeown-Longo, P.J.; Higgins, P.J. TGF-β1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. J. Cell Sci. 2001, 114, 3905–3914. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, C.E.; Higgins, S.P.; Kutz, S.M.; Higgins, P.J. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp60c-src/MEK-dependent. J. Cell Physiol. 2005, 204, 236–246. [Google Scholar] [CrossRef] [PubMed]
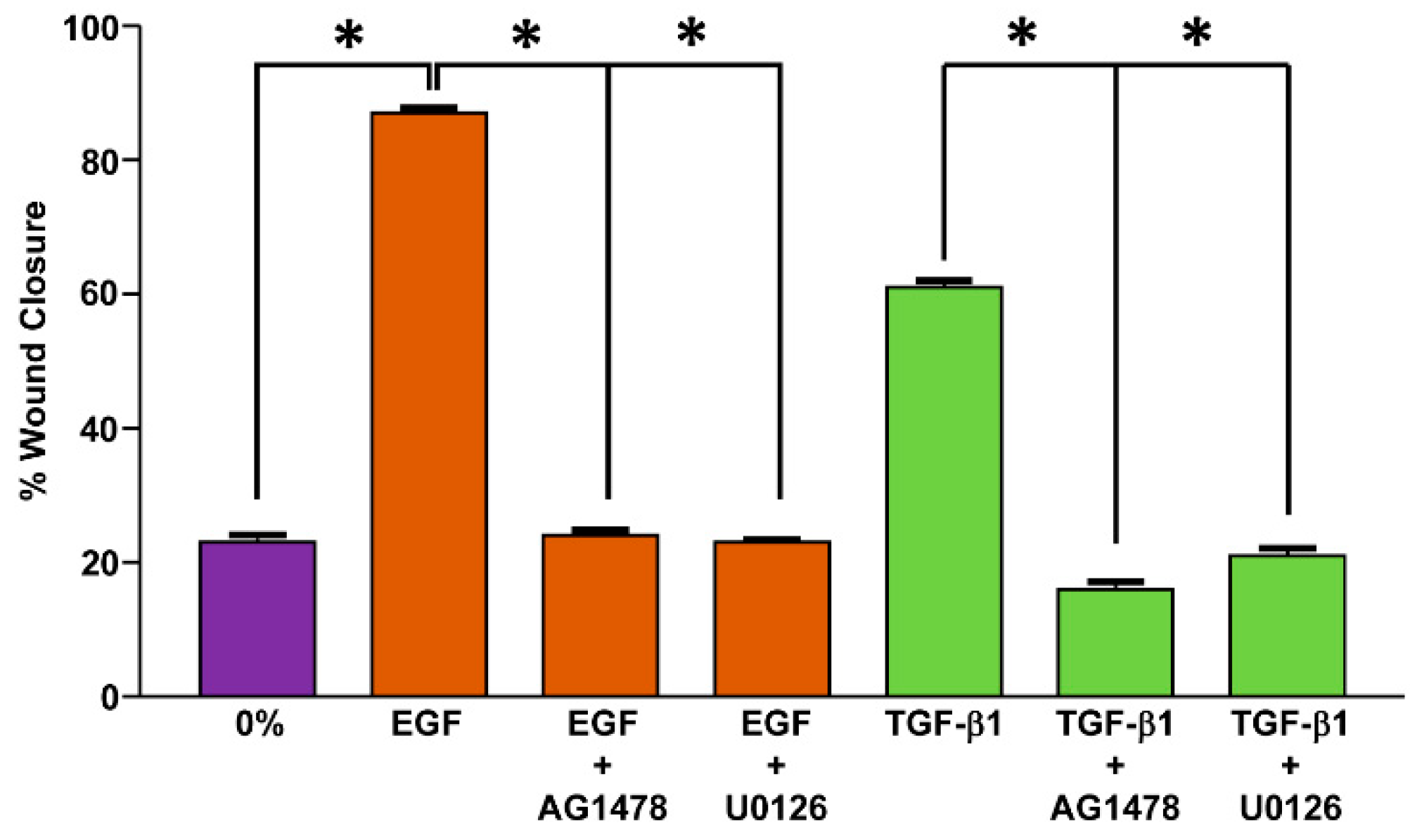
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef] [PubMed]
- Kutz, S.M.; Higgins, C.E.; Samarakoon, R.; Higgins, S.P.; Allen, R.R.; Qi, L.; Higgins, P.J. TGF-β1-induced PAI-1 expression is E box/USF-dependent and requires EGFR signaling. Exp. Cell Res. 2006, 312, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liu, Y.F.; Yang, L.; Wei, Q.; Zeng, H. Mechanism and clinical significance of the prothrombotic state in patients with essential hypertension. Clin. Cardiol. 2010, 33, E81–E86. [Google Scholar] [CrossRef] [PubMed]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Herszényi, L.; Plebani, M.; Cardin, R.; Carraro, P.; De Paoli, M.; Roveroni, G.; Naccarato, R.; Farinati, F. The role of urokinase-type plasminogen activator and its inhibitor PAI-1 in gastric cancer. Acta Physiol. Hung. 1995, 83, 213–221. [Google Scholar] [PubMed]
- Mashiko, S.; Kitatani, K.; Toyoshima, M.; Ichimura, A.; Dan, T.; Usui, T.; Ishibashi, M.; Shigeta, S.; Nagase, S.; Miyata, T.; et al. Inhibition of plas-minogen activator inhibitor-1 is a potential therapeutic strategy in ovarian cancer. Cancer Biol. Ther. 2015, 16, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Palmirotta, R.; Ferroni, P.; Savonarola, A.; Martini, F.; Ciatti, F.; Laudisi, A.; Sini, V.; Del Monte, G.; Guadagni, F.; Roselli, M. Prognostic value of pre-surgical plasma PAI-1 (plasminogen activator inhibitor-1) levels in breast cancer. Thromb. Res. 2009, 124, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.; Xu, M.; Sheng, W. Effects of CAF-derived microRNA on tumor biology and clinical applications. Cancers 2021, 13, 3160. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Pavón, M.A.; Arroyo-Solera, I.; Téllez-Gabriel, M.; León, X.; Virós, D.; López, M.; Gallardo, A.; Céspedes, M.V.; Casanova, I.; López-Pousa, A.; et al. Enhanced cell migration and apoptosis resistance may underlie the association between high SERPINE1 expression and poor outcome in head and neck carcinoma patients. Oncotarget 2015, 6, 29016–29033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekarda, H.; Schmitt, M.; Ulm, K.; Wenninger, A.; Vogelsang, H.; Becker, K.; Roder, J.D.; Fink, U.; Siewert, J.R. Prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in completely resected gastric cancer. Cancer Res. 1994, 54, 2900–2907. [Google Scholar] [PubMed]
- Annecke, K.; Schmitt, M.; Euler, U.; Zerm, M.; Paepke, D.; Paepke, S.; von Minckwitz, G.; Thomssen, C.; Harbeck, N. UPA and PAI-1 in breast cancer: Review of their clinical utility and current validation in the prospective NNBC-3 trial. Adv. Clin. Chem. 2008, 45, 31–45. [Google Scholar] [PubMed]
- Ahluwalia, P.; Ahluwalia, M.; Mondal, A.K.; Sahajpal, N.; Kota, V.; Rojiani, M.V.; Rojiani, A.M.; Kolhe, R. Prognostic and therapeutic implications of extracellular matrix associated gene signature in renal clear cell carcinoma. Sci. Rep. 2021, 11, 7561. [Google Scholar] [CrossRef] [PubMed]
- Kasza, A.; Kowanetz, M.; Poślednik, K.; Witek, B.; Kordula, T.; Koj, A. Epidermal growth factor and pro-inflammatory cytokines regulate the expression of components of plasminogen activation system in U373-MG astrocytoma cells. Cytokine 2001, 16, 187–190. [Google Scholar] [CrossRef]
- Lucore, C.L.; Fau, F.S.; Wun, T.C.; Sobel, B.E.; Billadello, J.J. Regulation of the expression of type 1 plasminogen activator inhibitor in HEP G2 cells by epidermal growth factor. J. Biol. Chem. 1988, 263, 15845–15848. [Google Scholar] [CrossRef]
- Wilkins-Port, C.E.; Higgins, P.J. Regulation of extracellular matrix remodeling following transforming growth factor-β1/epidermal growth factor-stimulated epithelial-mesenchymal transition in human premalignant keratinocytes. Cells Tissues Organs 2007, 185, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.S.; Marwitz, S.; Heilenkötter, U.; Schumm, W.; Behrens, O.; Simon, R.; Reck, M.; Vollmer, E.; Goldmann, T. Transforming growth factor-beta signaling leads to uPA/PAI-1 activation and metastasis: A study on human breast cancer tissues. Pathol. Oncol. Res. 2014, 20, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chakraborty, C.; Lala, P.K. Restoration of TGF-β regulation of plasminogen activator inhibitor-1 in Smad3-restituted human choriocarcinoma cells. Boichem. Biophys. Res. Commun. 2002, 294, 1079–1086. [Google Scholar] [CrossRef]
- Magnussen, S.N.; Hadler-Olsen, E.; Costea, D.E.; Berg, E.; Jacobsen, C.C.; Mortensen, B.; Salo, T.; Martinez-Zubiaurre, I.; Winberg, J.O.; Uhlin-Hansen, L.; et al. Cleavage of the urokinase receptor (uPAR) on oral cancer cells: Regulation by transforming growth factor-β1 (TGF-β1) and potential effects on migration and invasion. BMC Cancer 2017, 17, 350. [Google Scholar] [CrossRef]
- Humbert, L.; Lebrun, J.-J. TGF-β inhibits human cutaneous melanoma cell migration and invasion through regulation of the plasminogen activator system. Cell Signal. 2013, 25, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Konrad, L.; Scheiber, J.A.; Schwarz, L.; Schrader, A.J.; Hofmann, R. TGF-β1 and TGF-β2 strongly enhance the secretion of plasminogen activator inhibitor-1 and matrix metalloproteinase-9 of the human prostate cancer cell line PC-3. Regul. Pept. 2009, 155, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Albo, D.; Berger, D.H.; Vogel, J.; Tuszynski, G.P. Thrombospondin-1 and transforming growth factor β-1 upregulate plasminogen activator inhibitor type 1 in pancreatic cancer. J. Gastrointest. Surg. 1999, 3, 411–417. [Google Scholar] [CrossRef]
- Paugh, B.S.; Paugh, S.W.; Bryan, L.; Kapitonov, D.; Wilczynska, K.M.; Gopalan, S.M.; Rokita, H.; Milstien, S.; Spiegel, S.; Kordula, T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008, 22, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyrzykowska, P.; Stalińska, K.; Wawro, M.; Kochan, J.; Kasza, A. Epidermal growth factor regulates PAI-1 expression via activation of the transcription factor Elk-1. Biochim. Biophys. Acta 2010, 1799, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Zieba, A.; Vasilaki, E.; Herrera Hidalgo, C.; Söderberg, O.; Koinuma, D.; Miyazono, K.; Heldin, C.H.; Landegren, U.; Ten Dijke, P.; et al. Specific interactions between Smad proteins and AP-1 components determine TGFβ-induced breast cancer cell invasion. Oncogene 2013, 32, 3606–3615. [Google Scholar] [CrossRef] [Green Version]
- Overstreet, J.M.; Samarakoon, R.; Meldrum, K.K.; Higgins, P.J. Redox control of p53 in the transcriptional regulation of TGF-β1 target genes through SMAD cooperativity. Cell Signal. 2014, 26, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Overstreet, J.M.; Samarakoon, R.; Cardona-Grau, D.; Goldschmeding, R.; Higgins, P.J. Tumor suppressor ataxia telangiectasia mutated functions downstream of TGF-β1 in orchestrating profibrotic responses. FASEB J. 2015, 29, 1258–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.Y.; Wang, Y.; Su, J.; Huang, G.X.; Cao, D.-M.; Qu, S.; Lu, J. The mechanism and significance of synergistic induction of the expression of plasminogen activator inhibitor-1 by glucocorticoid and transforming growth factor beta in human ovarian cancer cells. Mol. Cell Endocrinol. 2015, 407, 37–45. [Google Scholar] [CrossRef]
- Wilkins-Port, C.E.; Higgins, C.E.; Freytag, J.; Higgins, S.P.; Carlson, J.A.; Higgins, P.J. PAI-1 is a critical upstream regulator of the TGF-β1/EGF-induced invasive phenotype in mutant p53 human cutaneous squamous cell carcinoma. J. Biomed. Biotechnol. 2007, 2007, 85208. [Google Scholar] [CrossRef] [Green Version]
- Freytag, J.; Wilkins-Port, C.H.; Higgins, C.E.; Carlson, J.A.; Noel, A.; Foidart, J.-M.; Higgins, S.P.; Samarakoon, R.; Higgins, P.J. PAI-1 regulates the invasive phenotype in human cutaneous squamous cell carcinoma. J. Oncol. 2009, 2009, 963209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahovac, J.; Wells, A. Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Lab. Investig. 2013, 94, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubriac, J.; Han, S.; Grahovac, J.; Smith, E.; Hosein, A.; Buchanan, M.; Basik, M.; Boucher, Y. The crosstalk between breast carcinoma-associated fibroblasts and cancer cells promotes RhoA-dependent invasion via IGF-1 and PAI-1. Oncotarget 2017, 9, 10375–10387. [Google Scholar] [CrossRef] [Green Version]
- Sitaram, R.T.; Mallikarjuna, P.; Landström, M.; Ljungberg, B. Transforming growth factor-β promotes aggressiveness and invasion of clear cell renal cell carcinoma. Oncotarget 2016, 7, 35917–35931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
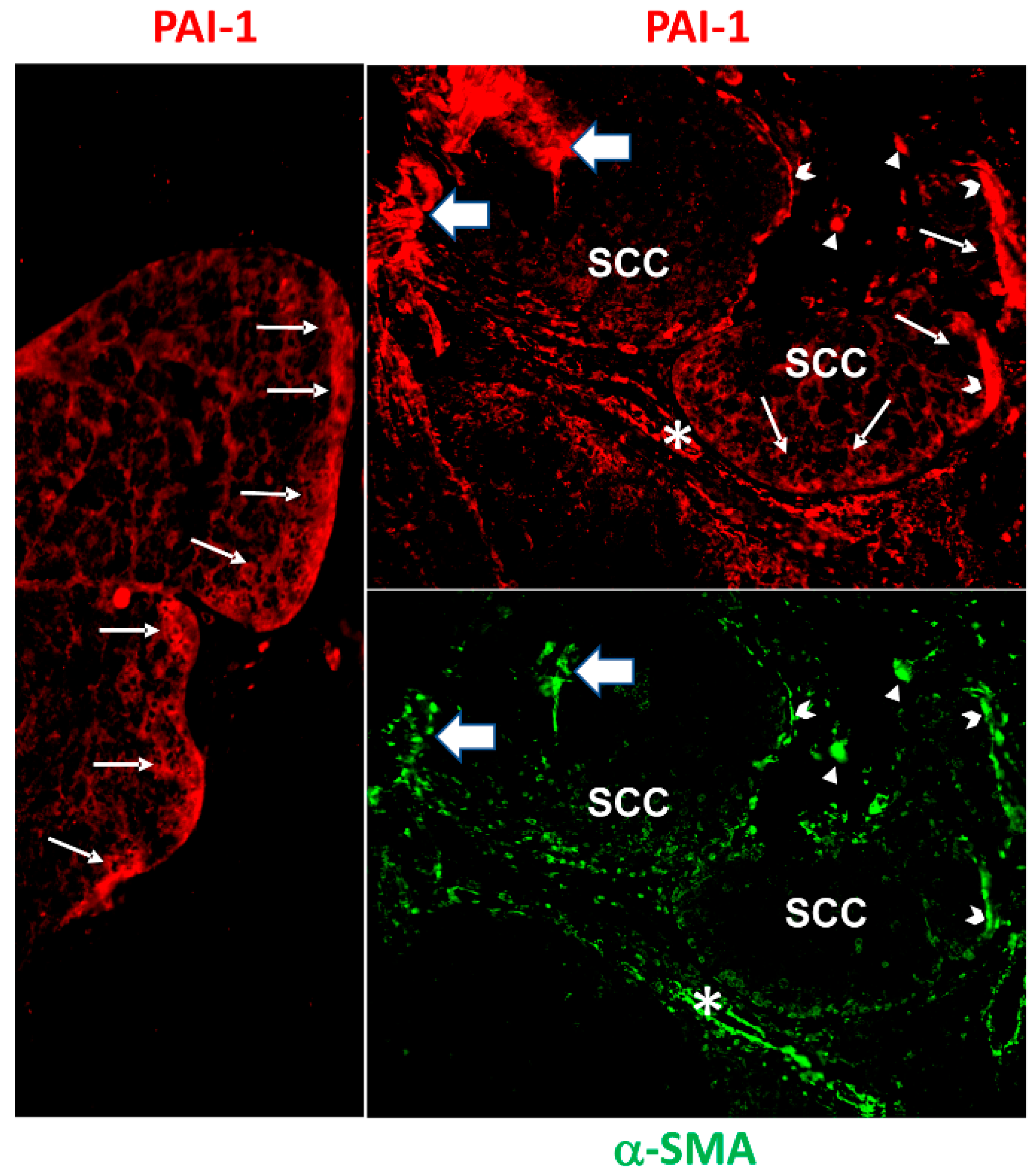
- Sakamoto, H.; Koma, Y.-I.; Higashino, N.; Kodama, T.; Tanigawa, K.; Shimizu, M.; Fujikawa, M.; Nishio, M.; Shigeoka, M.; Kakeji, Y.; et al. PAI-1 derived from cancer-associated fibroblasts in esophageal squamous cell carcinoma promotes the invasion of cancer cells and the migration of macrophages. Lab. Investig. 2021, 101, 353–368. [Google Scholar] [CrossRef]
- Wei, X.; Li, S.; He, J.; Du, H.; Liu, Y.; Yu, W.; Hu, H.; Han, L.; Wang, C.; Li, H.; et al. Tumor-secreted PAI-1 promotes breast cancer metastasis via the induction of adipocyte-derived collagen remodeling. Cell Commun. Signal. 2019, 17, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagordakis, E.; Sawazaki-Calone, I.; Macedo, C.C.; Carnielli, C.M.; de Oliveira, C.E.; Rodrigues, P.C.; Rangel, A.L.; Dos Santos, J.N.; Risteli, J.; Graner, E.; et al. Secretome profiling of oral squamous cell carcinoma-associated fibroblasts reveals organization and disassembly of extracellular matrix and collagen metabolic process signatures. Tumour Biol. 2016, 37, 9045–9057. [Google Scholar] [CrossRef]
- Czekay, R.-P.; Higgins, P.J. The SERPINE1/LRP1 axis at the crossroads of downstream signaling to cell motility. Trends Cell Mol. Biol. 2018, 13, 85–98. [Google Scholar]
- Tan, P.; Chen, H.; Huang, Z.; Huang, M.; Du, Y.; Li, T.; Chen, Z.; Liu, Y.; Fu, W. MMP25-AS1/hsa-miR-10a-5p/SERPINE1 axis as a novel prognostic biomarker associated with immune cell infiltration in kirc. Mol. Ther. Oncolytics 2021, 22, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Lu, K.; Fu, L. Prediction and analysis of novel key genes ITGAX, LAPTM5, SERPINE1 in clear cell renal cell carcinoma through bioinformatics analysis. PeerJ 2021, 9, e11272. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wan, H.; Zhu, M.; Feng, L.; Zhang, H.; Su, F. Discovery and validation of an epithelial-mesenchymal transition-based signature in gastric cancer by genomics and prognosis analysis. Biomed. Res. Int. 2021, 2021, 9026918. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Huang, M.; Tang, S.; Wang, Q.; Hu, P.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S.; Zhang, L. Plasminogen activator inhibitor (PAI-1) trap3, an exocellular peptide inhibitor of PAI-1, attenuates the rearrangement of F-actin and migration of cancer cells. Exp. Cell Res. 2020, 391, 111987. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.; Malinowsky, K.; Berg, D.; Schragner, K.; Schuster, T.; Walch, A.; Bronger, H.; Höfler, H.; Becker, K.F. Signalling networks associated with urokinase-type plasminogen activator (uPA) and its inhibitor PAI-1 in breast cancer tissues: New insights from protein microarray analysis. J. Pathol. 2011, 223, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Wang, R.; Kong, D.; Gao, Q.; Xu, C. Identification of potential core genes in gastric cancer using bioinformatics analysis. J. Gastrointest. Oncol. 2021, 12, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Quan, Q.; Xiong, X.; Wu, S.; Yu, M. Identification of autophagy-related prognostic signature and analysis of immune cell infiltration in low-grade gliomas. Biomed. Res. Int. 2021, 2021, 7918693. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Li, H.T.; Yu, J.P.; Han, X.P.; Xu, Z.P.; Li, Y.M.; Jiao, Z.Y.; Liu, H.B. A competing endogenous RNA network reveals novel potential lncRNA, miRNA, and mRNA biomarkers in the prognosis of human colon adenocarcinoma. J. Surg. Res. 2019, 235, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X.; Tian, Y.; Zhu, G.; Qin, Y.; Chen, X.; Pi, L.; Wei, M.; Liu, G.; Li, Z.; et al. Six-gene signature for predicting survival in patients with head and neck squamous cell carcinoma. Aging 2020, 12, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Liu, D.; Zhu, J.; Zhang, T. Common gene signatures and key pathways in hypopharyngeal and esophageal squamous cell carcinoma: Evidence from bioinformatic analysis. Medicine 2020, 99, e22434. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.; Ning, W.; Zhang, H.; Yu, C. Identification of two core genes in glioblastomas with different isocitrate dehydrogenase mutation status. Mol. Biol. Rep. 2020, 47, 7477–7488. [Google Scholar] [CrossRef]
- Tang, X.Z.; Zhou, X.G.; Zhang, X.G.; Li, G.S.; Chen, G.; Dang, Y.W.; Huang, Z.G.; Li, M.X.; Liang, Y.; Yao, Y.X.; et al. The clinical significance of interleukin 24 and its potential molecular mechanism in laryngeal squamous cell carcinoma. Cancer Biomark. 2020, 29, 111–124. [Google Scholar] [CrossRef]
- Xiao, Y. Construction of a circRNA-miRNA-mRNA network to explore the pathogenesis and treatment of pancreatic ductal adenocarcinoma. J. Cell Biochem. 2020, 121, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, Z.; Zhao, Y.; Zhang, Q.; Wu, X.; Miao, B.; Cao, J.; Fei, S. FN1, SPARC, and SERPINE1 are highly expressed and significantly related to a poor prognosis of gastric adenocarcinoma revealed by microarray and bioinformatics. Sci. Rep. 2019, 9, 7827. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhang, Y.; Yang, J. Identification of potentially functional circRNA-miRNA-mRNA regulatory network in gastric carcinoma using bioinformatics analysis. Med. Sci. Monit. 2019, 25, 8777–8796. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chi, W.; Cao, H.; Cui, W.; Meng, W.; Guo, W.; Wang, B. Screening and clinical significance of tumor markers in head and neck squamous cell carcinoma through bioinformatics analysis. Mol. Med. Rep. 2019, 19, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Xu, J.; Ru, L.; Zhu, W. Identification of pivotal genes associated with the prognosis of gastric carcinoma through integrated analysis. Biosci. Rep. 2021, 41, BSR20203676. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hu, X.; Dai, B.; Wang, Q.; Wang, H. Bioinformatics analysis of laryngeal squamous cell carcinoma: Seeking key candidate genes and pathways. PeerJ 2021, 9, e11259. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Du, G.; Zhao, R.; Yang, W.; Li, C.; Huang, J.; Wen, Z.; Li, H.; Zhang, B. Identification and validation of a hypoxia-related prognostic signature in clear cell renal cell carcinoma patients. Medicine 2021, 100, e27374. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Miyasaka, Y.; Ohuchida, K.; Okumura, T.; Zheng, B.; Torata, N.; Fujita, H.; Nabae, T.; Manabe, T.; Shimamoto, M.; et al. Calpain inhibitor calpeptin suppresses pancreatic cancer by disrupting cancer-stromal interactions in a mouse xenograft model. Cancer Sci. 2016, 107, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y.; et al. Inhibition of PAI-1 limits chemotherapy resistance in lung cancer through suppressing myofibroblast characteristics of cancer-associated fibroblasts. J. Cell Mol. Med. 2019, 23, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.R.; Eisen, M.B.; Ross, D.T.; Schuler, G.; Moore, T.; Lee, J.C.; Trent, J.M.; Staudt, L.M.; Hudson, J., Jr.; Boguski, M.S.; et al. The transcriptional program in the response of human fibroblasts to serum. Science 1999, 283, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of plasminogen activator inhibitor-1 attenuates transforming growth factor-β-dependent epithelial mesenchymal transition and differentiation of fibroblasts to myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef] [Green Version]
- Tsuge, M.; Osaki, M.; Sasaki, R.; Hirahata, M.; Okada, F. SK-216, a novel inhibitor of plasminogen activator inhibitor-1, suppresses lung metastasis of human osteosarcoma. Int. J. Mol. Sci. 2018, 19, 736. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Hattori, N.; Senoo, T.; Akita, S.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Kohno, N. SK-216, an inhibitor of plasminogen activator inhibitor-1, limits tumor progression and angiogenesis. Mol. Cancer Ther. 2013, 12, 2378–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Shen, H.; Zhu, L.; Zhao, F.; Shu, Y. Plasminogen activator inhibitor 1 promotes immunosuppression in human non-small cell lung cancers by enhancing TGF-β1 expression in macrophage. Cell Physiol. Biochem. 2017, 44, 2201–2211. [Google Scholar] [CrossRef]
- Tseng, Y.J.; Lee, C.H.; Chen, W.Y.; Yang, J.L.; Tzeng, H.T. Inhibition of PAI-1 blocks PD-L1 endocytosis and improves the response of melanoma cells to immune checkpoint blockade. J. Investig. Dermatol. 2021, 141, 2690–2698.e2696. [Google Scholar] [CrossRef]
- Ohuchi, K.; Kambayashi, Y.; Hidaka, T.; Fujimura, T. Plasminogen activating inhibitor-1 might predict the efficacy of anti-PD1 antibody in advanced melanoma patients. Front. Oncol. 2021, 11, 798385. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Liu, K.; Liu, J.H.; Jiang, X.L.; Wang, X.L.; Chen, Y.Z.; Li, S.G.; Zou, H.; Pang, L.J.; Liu, C.X.; et al. CD163 as a marker of M2 macrophage, contribute to predict aggressiveness and prognosis of Kazakh esophageal squamous cell carcinoma. Oncotarget 2017, 8, 21526–21538. [Google Scholar] [CrossRef] [Green Version]
- Kubala, M.H.; Punj, V.; Placencio-Hickok, V.R.; Fang, H.; Fernandez, G.E.; Sposto, R.; DeClerck, Y.A. Plasminogen activator inhibitor-1 promotes the recruitment and polarization of macrophages in cancer. Cell Rep. 2018, 25, 2177–2191.e2177. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.W.; Olman, M.; Benveniste, E.N. Cxcl12-mediated induction of plasminogen activator inhibitor-1 expression in human CXCR4 positive astroglioma cells. Biol. Pharm. Bull. 2009, 32, 573–577. [Google Scholar] [CrossRef] [Green Version]
- Elokdah, H.; Abou-Gharbia, M.; Hennan, J.K.; McFarlane, G.; Mugford, C.P.; Krishnamurthy, G.; Crandall, D.L. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: Design, synthesis, and preclinical characterization. J. Med. Chem. 2004, 47, 3491–4394. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef]
- Correa, R.J.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis 2012, 33, 49–58. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemo-resistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef]
- Musa, F.; Alard, A.; David-West, G.; Curtin, J.P.; Blank, S.V.; Schneider, R.J. Dual mTORC1/2 inhibition as a novel strategy for the resensitization and treatment of platinum-resistant ovarian cancer. Mol. Cancer Ther. 2016, 15, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Uno, S.; Watanabe, Y.; Arakawa, K.; Nakagawa, S. Expression of nerve growth factor-induced type 1 plasminogen activator inhibitor (PAI-1) mRNA is inhibited by genistein and wortmannin. Neuroreport 2000, 11, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
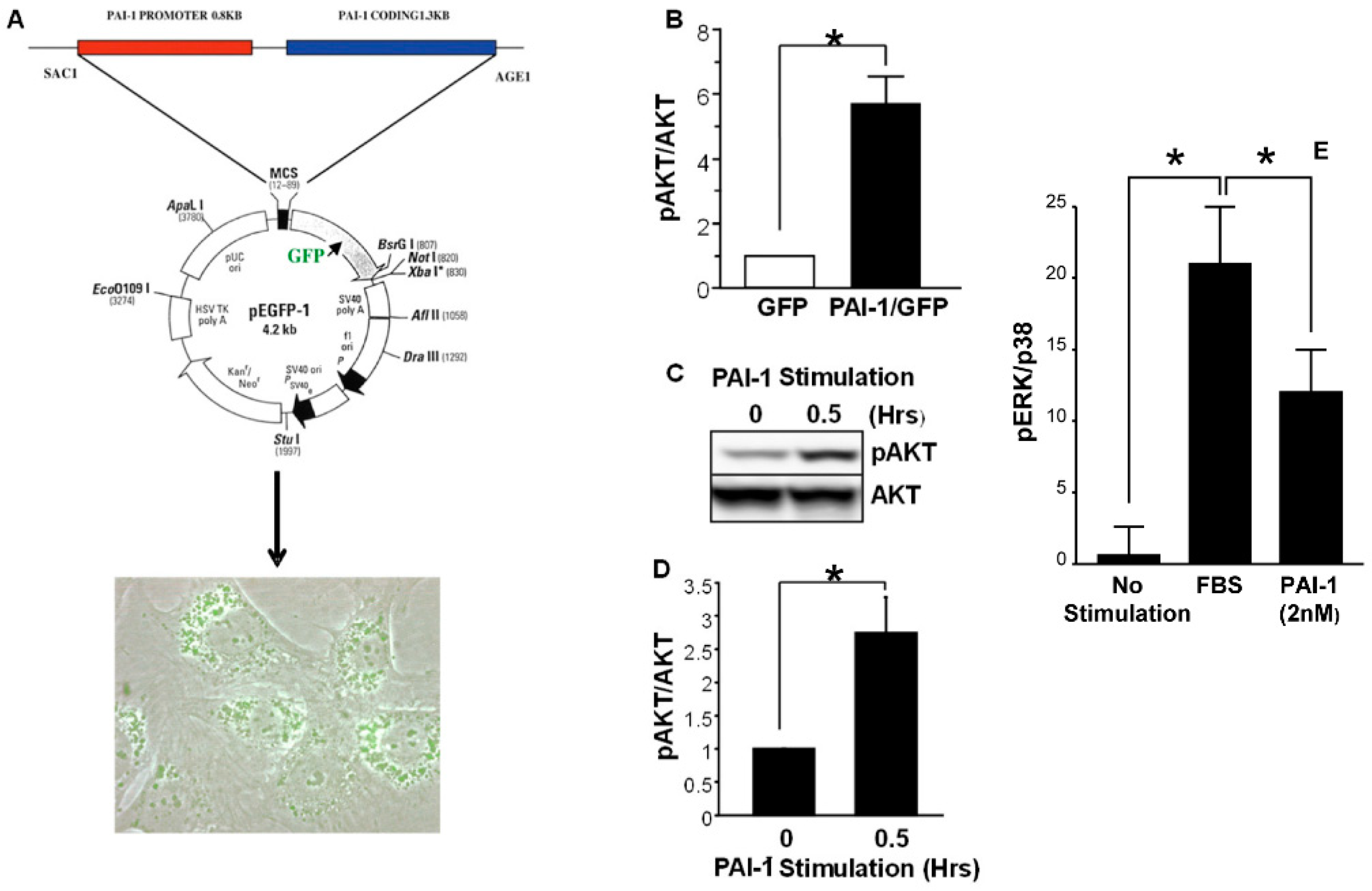
- Rømer, M.U.; Larsen, L.; Offenberg, H.; Brünner, N.; Lademann, U.A. Plasminogen activator inhibitor 1 protects fibrosarcoma cells from etoposide-induced apoptosis through activation of the PI3K/Akt cell survival pathway. Neoplasia 2008, 10, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Li, J.; Li, Q.; Wang, X.; Medikonda, R.; Zhao, T.; Li, T.; Ma, H.; Yi, L.; Liu, P.; et al. ACT001 reduces the expression of PD-L1 by inhibiting the phosphorylation of STAT3 in glioblastoma. Theranostics 2020, 10, 5943–5956. [Google Scholar] [CrossRef]
- Xi, X.; Liu, N.; Wang, Q.; Chu, Y.; Yin, Z.; Ding, Y.; Lu, Y. Act001, a novel PAI-1 inhibitor, exerts synergistic effects in combination with cisplatin by inhibiting PI3K/AKT pathway in glioma. Cell Death Dis. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, T.M.; Higgins, S.P.; Archambeault, J.; Higgins, C.E.; Ginnan, R.G.; Singer, H.; Higgins, P.J. A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage. Cell Signal. 2015, 27, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Fersching, D.M.; Nagel, D.; Siegele, B.; Salat, C.; Heinemann, V.; Holdenrieder, S.; Stoetzer, O.J. Apoptosis-related biomarkers sFAS, MIF, ICAM-1 and PAI-1 in serum of breast cancer patients undergoing neoadjuvant chemotherapy. Anticancer Res. 2012, 32, 2047–2058. [Google Scholar] [PubMed]
- Fen Li, C.; Kandel, C.; Baliko, F.; Nadesan, P.; Brünner, N.; Alman, B.A. Plasminogen activator inhibitor-1 (PAI-1) modifies the formation of aggressive fibromatosis (desmoid tumor). Oncogene 2005, 24, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Placencio, V.R.; DeClerck, Y.A. Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J. Natl. Cancer Inst. 2012, 104, 1470–1484. [Google Scholar] [CrossRef] [Green Version]
- Bajou, K.; Peng, H.; Laug, W.E.; Maillard, C.; Noel, A.; Foidart, J.M.; Martial, J.A.; DeClerck, Y.A. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 2008, 14, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, R.; Kaji, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Kitade, M.; Takaya, H.; Aihara, Y.; Douhara, A.; Asada, K.; et al. Novel oral plasminogen activator inhibitor-1 inhibitor TM5275 attenuates hepatic fibrosis under metabolic syndrome via suppression of activated hepatic stellate cells in rats. Mol. Med. Rep. 2020, 22, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, A.; Matsumoto, S.; Suzuki, S.; Dan, T.; Yamaki, S.; Sato, Y.; Kiyomoto, H.; Ishii, N.; Okada, K.; Matsuo, O.; et al. A small molecule inhibitor to plasminogen activator inhibitor 1 inhibits macrophage migration. Arterioslcer. Thromb. Vasc. Biol. 2013, 33, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Dong, Q.; Hu, S.; Cai, J.; Zhang, W.; Sun, J.; Wang, T.; Xie, J.; He, H.; Xing, J.; et al. Proteomic analysis of the proteins that are associated with the resistance to paclitaxel in human breast cancer cells. Mol. Biosyst. 2014, 10, 294–303. [Google Scholar] [CrossRef]
- Panayotopoulou, E.G.; Müller, A.K.; Börries, M.; Busch, H.; Hu, G.; Lev, S. Targeting of apoptotic pathways by smac or bh3 mimetics distinctly sensitizes paclitaxel-resistant triple negative breast cancer cells. Oncotarget 2017, 8, 45088–45104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lei, L.; Jing, D. Knockdown of SERPINE1 reverses resistance of triple-negative breast cancer to paclitaxel via suppression of VEGFA. Oncol. Rep. 2020, 44, 1875–1884. [Google Scholar] [CrossRef]
- Providence, K.M.; Higgins, S.P.; Mullen, A.; Battista, A.; Samarakoon, R.; Higgins, C.E.; Wilkins-Port, C.E.; Higgins, P.J. SERPINE1 (PAI-1) is deposited into keratinocyte migration “trails” and required for optimal monolayer wound repair. Arch. Dermatol. Res. 2008, 300, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, S.P.; Samarakoon, R.; Higgins, C.E.; Freytag, J.; Wilkins-Port, C.E.; Higgins, P.J. TGF-β1-induced expression of the anti-apoptotic PAI-1 protein requires EGFR signaling. Cell Commun. Insights 2009, 2, 1–11. [Google Scholar] [PubMed]
- Pan, J.-X.; Qu, F.; Wang, F.-F.; Xu, J.; Mu, L.-S.; Ye, L.-Y.; Li, J.-J. Aberrant SERPINE1 DNA methylation is involved in carboplatin induced epithelial-mesenchymal transition in epithelial ovarian cancer. Arch. Gynecol. Obstet. 2017, 296, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ono, Y.J.; Kanemura, M.; Tanaka, T.; Hayashi, M.; Terai, Y.; Ohmichi, M. Hepatocyte growth factor secreted by ovarian cancer cells stimulates peritoneal implantation via the mesothelial-mesenchymal transition of the peritoneum. Gynecol. Oncol. 2015, 139, 345–354. [Google Scholar] [CrossRef]
- Peng, Y.; Kajiyama, H.; Yuan, H.; Nakamura, K.; Yoshihara, M.; Yokoi, A.; Fujikake, K.; Yasui, H.; Yoshikawa, N.; Suzuki, S.; et al. PAI-1 secreted from metastatic ovarian cancer cells triggers the tumor-promoting role of the mesothelium in a feedback loop to accelerate peritoneal dissemination. Cancer Lett. 2019, 442, 181–192. [Google Scholar] [CrossRef]
- Agarwal, A.; Tressel, S.L.; Kaimal, R.; Balla, M.; Lam, F.H.; Covic, L.; Kuliopulos, A. Identification of a metalloprotease-chemokine signaling system in the ovarian cancer microenvironment: Implications for antiangiogenic therapy. Cancer Res. 2010, 70, 5880–5890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikuła-Pietrasik, J.; Uruski, P.; Szubert, S.; Moszyński, R.; Szpurek, D.; Sajdak, S.; Tykarski, A.; Książek, K. Biochemical composition of malignant ascites determines high aggressiveness of undifferentiated ovarian tumors. Med. Oncol. 2016, 33, 94. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.-P.; Higgins, C.E.; Archambeault, J.; Higgins, S.P.; Simone, T.M.; Higgins, P.J. The small molecule PAI-1 functional inhibitor Tiplaxtinin attenuates vascular smooth muscle cell migration in vitro and neointimal hyperplasia/fibrosis in vivo. In Carotid Artery Disease; Avid Science: Dallas, TX, USA, 2017; pp. 2–40. [Google Scholar]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2019, 8, 64. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-dimensional culture system of cancer cells combined with biomaterials for drug screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Cao, H.; Cheng, H.S.; Wang, J.K.; Tan, S.N.; Tay, C.Y. A 3D physio-mimetic interpenetrating network-based platform to decode the pro and anti-tumorigenic properties of cancer-associated fibroblasts. Acta Biomater. 2021, 132, 448–460. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. A cancer invasion model combined with cancer-associated fibroblast aggregates incorporating geltain hydrogen microspheres containing a p53 inhibitor. Tissue Eng. Part C Methods 2019, 25, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Brancato, V.; Gioiella, F.; Imparato, G.; Guamieri, D.; Uriciuolo, F.; Netti, P.A. 3D breast cancer microtissue reveals the role of tumor microenvironment on the transport and efficacy of free-doxorubicin in vitro. Acta Biomater. 2018, 75, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Kay, E.J.; Zanivan, S. Metabolic pathways fuelling protumourigenic cancer-associated fibroblast functions. Curr. Opin. Syst. Biol. 2021, 29, 100377. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Yoon, J.-H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse warburg effect and its therapeutic implication. J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomengerg, N.; Kitkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburgh effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Santi, A.; Caselli, A.; Ranaldi, F.; Paoli, P.; Mugnaioni, C.; Michelucci, E.; Cirri, P. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta 2015, 1853, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.-S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.X.; Cheng, Z.; Yang, X.; Xhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Prestell, R.G.; Howell, A.; Aquila, S. Metabolic programming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF−β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Zaal, E.A.; Berkers, C.R. The influence of metabolism on drug response in cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef] [PubMed]
- Pranzini, E.; Pardella, E.; Paoli, P.; Fendt, S.-M.; Taddei, M.L. Metabolic reprogramming in anti-cancer drug resistance: A focus on amino acids. Trends Cancer 2021, 7, 682–699. [Google Scholar] [CrossRef]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and furious: Lipid metabolism in antitumoral therapy response and resistance. Trends Cancer 2021, 7, 198. [Google Scholar] [CrossRef]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef] [PubMed]
- Vander-Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, E.; Caiazza, C.; Mimmi, S.; Avagliano, A.; Iaccino, E.; Brusco, T.; Nisticò, N.; Maisano, D.; Aloisio, A.; Quinto, I.; et al. Metabolites profiling of melanoma interstitial fluids reveals uridine diphosphate as potent immune modulator capable of limiting tumor grwoth. Front. Cell Dev. Biol. 2021, 9, 730736. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Vander-Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Granato, G.; Ruocco, M.R.; Iaccarino, A.; Masone, S.; Cali, G.; Avagliano, A.; Russo, V.; Bellevicine, C.; Di Spigna, G.; Fiiume, G.; et al. Generation and analysis of spheroids from human primary skin myofibroblasts: An experimental system to study myofibroblasts deactivation. Cell Death Dis. 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Jaarsveld, M.T.; Helleman, J.; Berns, E.M.; Wiemer, E.A. MicroRNAs in ovarian cancer biology and therapy resistance. Int. J. Biochem. Cell Biol. 2010, 42, 1282–1290. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. MicroRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuberi, M.; Khan, I.; Mir, R.; Gandhi, G.; Ray, P.C.; Saxena, A. Utility of serum miR-125b as a diagnostic and prognostic indicator and its alliance with a panel of tumor suppressor genes in epithelial ovarian cancer. PLoS ONE 2016, 11, e0153902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat. Cell Biol. 2011, 14, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Melling, G.E.; Flannery, S.E.; Abidin, S.A.; Clemmens, H.; Prajapati, P.; Hinsley, E.E.; Hunt, S.; Catto, J.W.F.; Coletta, R.D.; Mellone, M.; et al. A miRNA-145/TGF-β1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis 2018, 39, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef]
- Botla, S.K.; Savant, S.; Jandaghi, P.; Bauer, A.S.; Mücke, O.; Moskalev, E.A.; Neoptolemos, J.P.; Costello, E.; Greenhalf, W.; Scarpa, A.; et al. Early epigenetic downregulation of microRNA-192 expression promotes pancreatic cancer progression. Cancer Res. 2016, 76, 4149–4159. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef]
- Yu, Z.; Baserga, R.; Chen, L.; Wang, C.; Lisanti, M.P.; Pestell, R.G. MicroRNA, cell cycle, and human breast cancer. Am. J. Pathol. 2010, 176, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Muth, M.; Theophile, K.; Hussein, K.; Jacobi, C.; Kreipe, H.; Bock, O. Hypoxia-induced down-regulation of microRNA-449a/b impairs control over targeted SERPINE1 (PAI-1) mRNA - A mechanism involved in SERPINE1 (PAI-1 overexpression). J. Transl. Med. 2010, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villardsen, S.B.; Bramsen, J.B.; Ostenfeld, M.S.; Wiklund, E.D.; Fristrup, N.; Gao, S.; Hansen, T.B.; Jensen, T.I.; Borre, M.; Ørntoft, T.F.; et al. The miR-143/-145 cluster regulates plasminogen activator inhibitor-1 in bladder cancer. Br. J. Cancer 2012, 106, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Osaki, M.; Takeshita, F.; Sugimoto, Y.; Kosaka, N.; Yamamoto, Y.; Yoshioka, Y.; Kobayashi, E.; Yamada, T.; Kawai, A.; Inoue, T.; et al. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol. Ther. 2011, 19, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Hirahayta, M.; Osaki, M.; Kanda, Y.; Sugimoto, Y.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Kawai, A.; Ito, H.; et al. PAI-1, a target gene of miR-143, regulates invasion and metastasis by upregulating MMP-13 expression of human osteosarcoma. Cancer Med. 2016, 5, 892–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-D.; Ma, L.; Zhu, Z. SERPINE1 as a cancer-promoting gene in gastric adenocarcinoma: Facilitates tumor cell proliferation, migration, and invasion by regulating EMT. J. Chemother. 2019, 31, 408–418. [Google Scholar] [CrossRef]
- Seker, F.; Cingoz, A.; Sur-Erdem, I.; Erguder, N.; Erkent, A.; Uyulur, F.; Selvan, M.E.; Gumus, Z.H.; Gonen, M.; Bayraktar, H.; et al. Identification of SERPINE1 as a regulator of glioblastoma cell dispersal with transcriptome profiling. Cancers 2019, 11, 1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd-Aziz, N.; Kamaruzman, N.I.; Poh, C.L. Development of microRNAs as potential therapeutics against cancer. J. Oncol. 2020, 2020, 8029721. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Function |
|---|---|
| FAP | ECM remodeling, protease activity |
| PDGFα,β | Receptor tyrosine kinase, angiogenesis, immune cell modulation |
| CD10 | Protease activity, cancer stemness, drug resistance |
| CD74 | Protein trafficking, chaperone activity, immunomodulation |
| GRP77 | Proinflammatory signaling, tumor stemness |
| α-SMA | Cell contractility, proliferation, desmoplasia |
| S100A4 | Tumor cell migration, proliferation, fibrosis, metastasis |
| PDPN | Cell motility |
| CD70 | T-cell regulation, tumor cell migration |
| Vimentin | Cytoskeletal organization, cell motility, tumor invasion |
| POSTN | ECM remodeling |
| CDK1 | Cell cycling |
| Mitogens | Proinvasion Factors |
|---|---|
| EGF | HGF |
| HGF | TGF-β |
| IGF-1 | CCL5 |
| SDF-1 | POSTN |
| FGF-1 | IL-6 |
| FGF-3 | IL-11 |
| Inflammatory Mediators | Stemness Factors |
| IL-1 | IL-6 |
| IL-6 | IL-8 |
| IL-8 | HGF |
| IL-11 | IGF-2 |
| LIF | POSTN |
| Various chemokines | |
| Proangiogenic Factors | Survival Factors |
| VEGFA | IGF-1 |
| SDF-1 | IGF-2 |
| FGF-2 | IL-6 |
| IL-8 | OPN |
| PDGF-C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czekay, R.-P.; Cheon, D.-J.; Samarakoon, R.; Kutz, S.M.; Higgins, P.J. Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets. Cancers 2022, 14, 1231. https://doi.org/10.3390/cancers14051231
Czekay R-P, Cheon D-J, Samarakoon R, Kutz SM, Higgins PJ. Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets. Cancers. 2022; 14(5):1231. https://doi.org/10.3390/cancers14051231
Chicago/Turabian StyleCzekay, Ralf-Peter, Dong-Joo Cheon, Rohan Samarakoon, Stacie M. Kutz, and Paul J. Higgins. 2022. "Cancer-Associated Fibroblasts: Mechanisms of Tumor Progression and Novel Therapeutic Targets" Cancers 14, no. 5: 1231. https://doi.org/10.3390/cancers14051231