
Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: From Epidemiology to Diagnostic Approach


Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology
3. Pathogenesis
3.1. Genetic Background
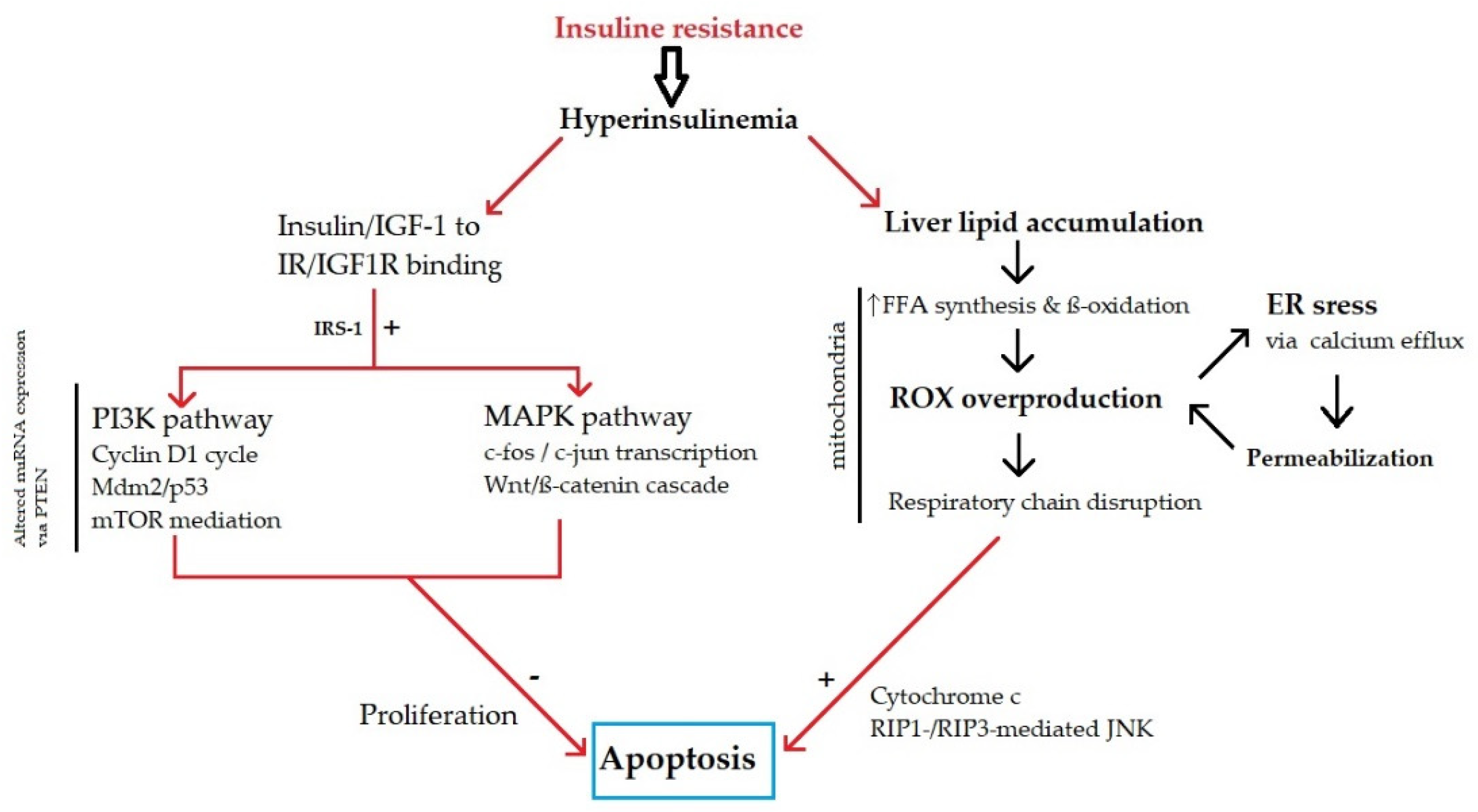
3.2. Metabolic Disbalance
3.3. The Role of Gut Microbiota
3.4. Immune-Mediated Mechanisms
4. Clinical Picture and Outcomes
4.1. Clinical Presentation of HCC in NAFLD
4.2. Clinical Outcomes of Patients with NAFLD-Related HCC
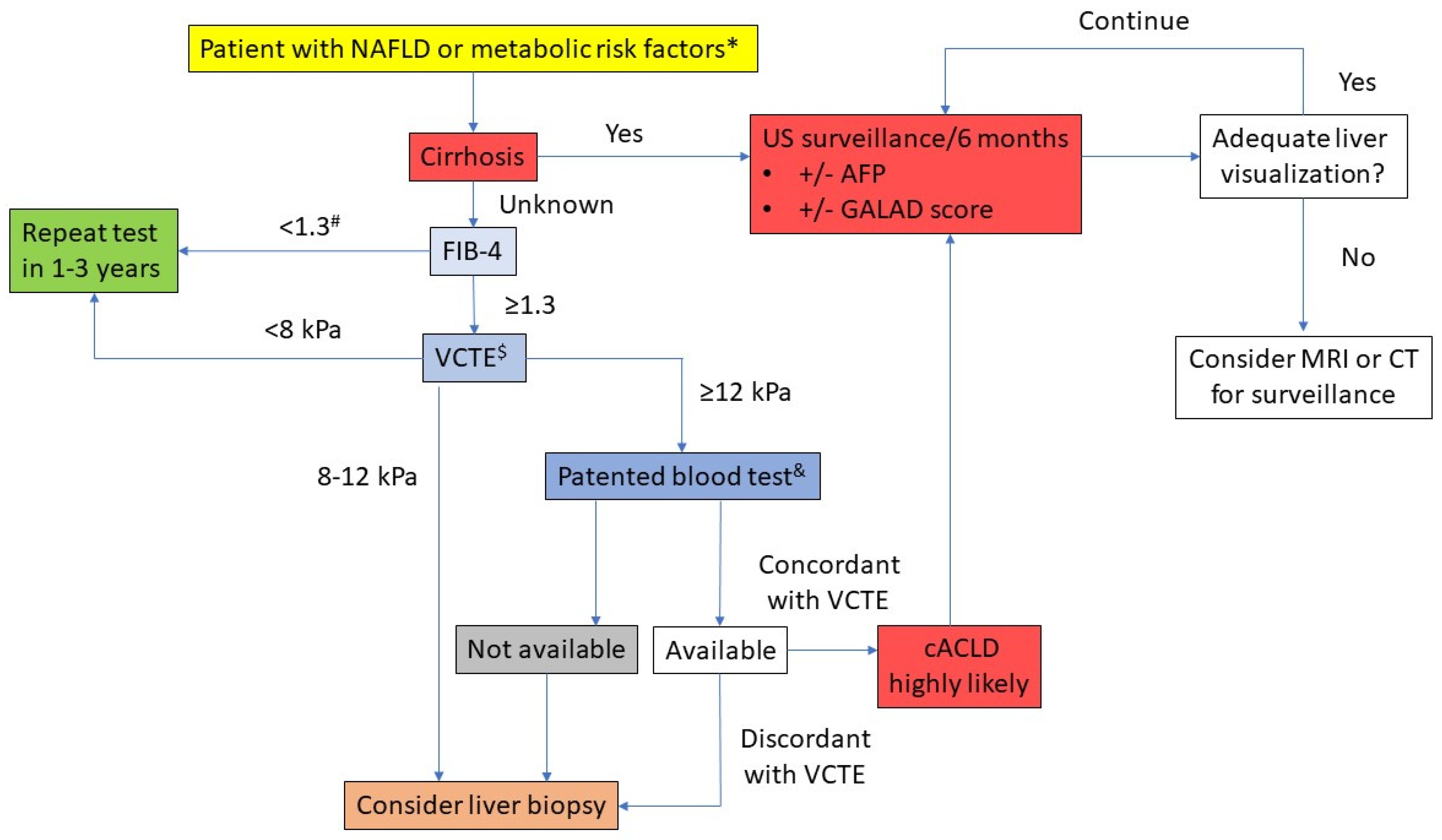
5. Diagnostic Approach
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Thursz, M.; Gual, A.; Lackner, C.; Mathurin, P.; Moreno, C.; Spahr, L.; Sterneck, M.; Cortez-Pinto, H. EASL clinical practice guidelines: Management of alcohol-related liver disease. J. Hepatol. 2018, 69, 154–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omata, M.; Cheng, A.-L.; Kokudo, N.; Kudo, M.; Lee, J.M.; Jia, J.; Tateishi, R.; Han, K.-H.; Chawla, Y.K.; Shiina, S.; et al. Asia–Pacific clinical practice guidelines on the management of hepatocellular carcinoma: A 2017 update. Hepatol. Int. 2017, 11, 317–370. [Google Scholar] [CrossRef] [Green Version]
- Shield, K.; Manthey, J.; Rylett, M.; Probst, C.; Wettlaufer, A.; Parry, C.D.; Rehm, J. National, regional, and global burdens of disease from 2000 to 2016 attributable to alcohol use: A comparative risk assessment study. Lancet Public Health 2020, 5, e51–e61. [Google Scholar] [CrossRef] [Green Version]
- World Health Organisation. Global Health Sector Strategy on Viral Hepatitis 2016–2021 Towards Ending Viral Hepatitis; WHO Press: Geneva, Switzerland, 2016. [Google Scholar]
- Flisiak, R.; Zarębska-Michaluk, D.; Frankova, S.; Grgurevic, I.; Hunyady, B.; Jarcuska, P.; Kupčinskas, L.; Makara, M.; Simonova, M.; Sperl, J.; et al. Is elimination of HCV in 2030 realistic in Central Europe. Liver Int. 2021, 41, 56–60. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 1 October 2021).
- Dufour, J.-F.; Scherer, R.; Balp, M.-M.; McKenna, S.J.; Janssens, N.; Lopez, P.; Pedrosa, M. The global epidemiology of Nonalcoholic Steatohepatitis (NASH) and associated risk factors–A targeted literature review. Endocr. Metab. Sci. 2021, 3, 100089. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Grgurevic, I.; Podrug, K.; Mikolasevic, I.; Kukla, M.; Madir, A.; Tsochatzis, E.A. Natural history of nonalcoholic fatty liver disease: Implications for clinical practice and an individualized approach. Can. J. Gastroenterol. Hepatol. 2020, 2020, e9181368. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Implications for liver transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Jothimani, D.; Danielraj, S.; Narasimhan, G.; Kaliamoorthy, I.; Rajakumar, A.; Palaniappan, K.; Palanisamy, S.; Rammohan, A.; Ramachandran, H.; Rajalingam, R.; et al. Non-alcoholic steatohepatitis: A rapidly increasing indication for liver transplantation in India. J. Clin. Exp. Hepatol. 2021. [Google Scholar] [CrossRef]
- Park, J.W.; Chen, M.; Colombo, M.; Roberts, L.R.; Schwartz, M.; Chen, P.J.; Kudo, M.; Johnson, P.; Wagner, S.; Orsini, L.S.; et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: The BRIDGE Study. Liver Int. 2015, 35, 2155–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Pais, R.; Fartoux, L.; Goumard, C.; Scatton, O.; Wendum, D.; Rosmorduc, O.; Ratziu, V. Temporal trends, clinical patterns and outcomes of NAFLD-related HCC in patients undergoing liver resection over a 20-year period. Aliment. Pharmacol. Ther. 2017, 46, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of metabolic traits on the risk of cirrhosis and hepatocellular cancer in nonalcoholic fatty liver disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Ahmed, F.; Mara, K.C.; Addissie, B.D.; Allen, A.M.; Gores, G.J.; Roberts, L.R. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 2020, 71, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969. [Google Scholar] [CrossRef] [PubMed]
- Yopp, A.C.; Choti, M.A. Non-alcoholic steatohepatitis-related hepatocellular carcinoma: A growing epidemic? Dig. Dis. 2015, 33, 642–647. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-Z.; Xia, H.H.-X.; Xin, Y.-N.; Lin, Z.-H.; Xuan, S.-Y. TM6SF2 E167K variant, a novel genetic susceptibility variant, contributing to nonalcoholic fatty liver disease. J. Clin. Transl. Hepatol. 2015, 3, 265. [Google Scholar] [PubMed] [Green Version]
- Ye, Q.; Qian, B.-X.; Yin, W.-L.; Wang, F.-M.; Han, T. Association between the HFE C282Y, H63D polymorphisms and the risks of non-alcoholic fatty liver disease, liver cirrhosis and hepatocellular carcinoma: An updated systematic review and meta-analysis of 5758 cases and 14,741 controls. PLoS ONE 2016, 11, e0163423. [Google Scholar] [CrossRef] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Thabet, K.; Chan, H.L.Y.; Petta, S.; Mangia, A.; Berg, T.; Boonstra, A.; Brouwer, W.P.; Abate, M.L.; Wong, V.W.S.; Nazmy, M.; et al. The membrane-bound O-acyltransferase domain-containing 7 variant rs641738 increases inflammation and fibrosis in chronic hepatitis B. Hepatology 2017, 65, 1840–1850. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.t.; Pan, K.; Chen, M.s.; Li, J.j.; Wang, H.; Zhao, J.j.; Sun, J.c.; Chen, Y.b.; Ma, H.q.; Wang, Q.j.; et al. Decreased expression of XPO4 is associated with poor prognosis in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 544–549. [Google Scholar] [CrossRef]
- Zain, S.M.; Mohamed, R.; Cooper, D.N.; Razali, R.; Rampal, S.; Mahadeva, S.; Chan, W.-K.; Anwar, A.; Rosli, N.S.M.; Mahfudz, A.S.; et al. Genome-wide analysis of copy number variation identifies candidate gene loci associated with the progression of non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e95604. [Google Scholar] [CrossRef]
- Liu, F.; Li, H.; Chang, H.; Wang, J.; Lu, J. Identification of hepatocellular carcinoma-associated hub genes and pathways by integrated microarray analysis. Tumori 2015, 101, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Han, T.; Muskhelishvili, L.; Fuscoe, J.C.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol. Nutr. Food Res. 2011, 55, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Takaki, Y.; Saito, Y.; Takasugi, A.; Toshimitsu, K.; Yamada, S.; Muramatsu, T.; Kimura, M.; Sugiyama, K.; Suzuki, H.; Arai, E.; et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci. 2014, 105, 1254–1260. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, M.F.; Wojczynski, M.K.; North, K.E.; Zhang, Q.; Province, M.A.; Carr, J.J.; Borecki, I.B. The ERLIN1-CHUK-CWF19L1 gene cluster influences liver fat deposition and hepatic inflammation in the NHLBI family heart study. Atherosclerosis 2013, 228, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.C.; Zhang, W.; Sehmi, J.; Li, X.; Wass, M.N.; Van der Harst, P.; Holm, H.; Sanna, S.; Kavousi, M.; Baumeister, S.E.; et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011, 43, 1131–1138. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef]
- Janku, F.; Kaseb, A.O.; Tsimberidou, A.M.; Wolff, R.A.; Kurzrock, R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget 2014, 5, 3012. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- De Conti, A.; Ortega, J.F.; Tryndyak, V.; Dreval, K.; Moreno, F.S.; Rusyn, I.; Beland, F.A.; Pogribny, I.P. MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget 2017, 8, 88517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Thematic review series: Lipotoxicity: Many roads to cell dysfunction and cell death: Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP 3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Bozaykut, P.; Sahin, A.; Karademir, B.; Ozer, N.K. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech. Ageing Dev. 2016, 157, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liao, J.-Z.; He, X.-X.; Li, P.-Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017, 8, 57707. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 2019, 84, 105803. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.S.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: A phase 2 randomized controlled trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Sanyal, A.; Lopez, P.; Lawitz, E.; Kim, W.; Huang, J.-F.; Andreone, P.; Goh, G.B.B.; Chen, Y.-C.; Ratziu, V.; Kim, Y.J.; et al. Tropifexor, a farnesoid X receptor agonist for the treatment of non-alcoholic steatohepatitis: Interim results based on baseline body mass index from first two parts of phase 2b study FLIGHT-FXR. J. Hepatol. 2019, 70, E796–E797. [Google Scholar] [CrossRef]
- Lucas, K.J.; Lopez, P.; Lawitz, E.J.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Goh, G.B.B.; Vierling, J.M.; Frias, J.P.; Whites, J. Safety and efficacy of tropifexor in patients with fibrotic nonalcoholic steatohepatitis: 48-week results from part C of the phase 2 flight-FXR study. In Proceedings of the Hepatology, Hoboken, NJ, USA, 25 November 2020; pp. 101A–102A. [Google Scholar]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [Green Version]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.L.; El-Nezami, H. Targeting gut microbiota in hepatocellular carcinoma: Probiotics as a novel therapy. Hepatobiliary Surg. Nutr. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukla, M.; Mazur, W.; Bułdak, R.J.; Żwirska-Korczala, K. Potential role of leptin, adiponectin and three novel adipokines—visfatin, chemerin and vaspin—in chronic hepatitis. Mol. Med. 2011, 17, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC–Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Augustyn, M.; Grys, I.; Kukla, M. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin. Exp. Hepatol. 2019, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Tao, X.; Wang, N.; Qin, W. Gut microbiota and hepatocellular carcinoma. Gastrointest. Tumors 2015, 2, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-B.; Gong, Y.-P.; Cheng, J.; Chu, Y.-W.; Xiong, S.-D. CXCL16 participates in pathogenesis of immunological liver injury by regulating T lymphocyte infiltration in liver tissue. World J. Gastroenterol. 2005, 11, 4979. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, 6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oellgaard, J.; Abitz Winther, S.; Schmidt Hansen, T.; Rossing, P.; Johan von Scholten, B. Trimethylamine N-oxide (TMAO) as a new potential therapeutic target for insulin resistance and cancer. Curr. Pharm. Des. 2017, 23, 3699–3712. [Google Scholar] [CrossRef]
- Yu, L.-X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Boursier, J.; Diehl, A.M. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog. 2015, 11, e1004559. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver—from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88. [Google Scholar] [CrossRef]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Sircana, A.; Paschetta, E.; Saba, F.; Molinaro, F.; Musso, G. Recent insight into the role of fibrosis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1745. [Google Scholar] [CrossRef] [Green Version]
- White, D.L.; Kanwal, F.; El–Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Reid, D.; Reyes, J.; McDonald, B.; Vo, T.; Reimer, R.; Eksteen, B. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS ONE 2016, 11, e0159524. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Nakashima, M.; Kinoshita, M.; Ikarashi, M.; Miyazaki, H.; Hanaka, H.; Imaki, J.; Seki, S. Activation and increase of radio-sensitive CD11b+ recruited Kupffer cells/macrophages in diet-induced steatohepatitis in FGF5 deficient mice. Sci. Rep. 2016, 6, 34466. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Csak, T.; Ganz, M.; Szabo, G. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2013, 28, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of hepatic progenitor cells in nonalcoholic fatty liver disease development: Cellular cross-talks and molecular networks. Int. J. Mol. Sci. 2013, 14, 20112–20130. [Google Scholar] [CrossRef] [Green Version]
- Leiser, Y.; Blumenfeld, A.; Haze, A.; Dafni, L.; Taylor, A.L.; Rosenfeld, E.; Fermon, E.; Gruenbaum-Cohen, Y.; Shay, B.; Deutsch, D. Localization, quantification, and characterization of tuftelin in soft tissues. Anat. Rec. (Hoboken) 2007, 290, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.L.; Shi, L.; Zhou, J.N.; Fu, C.J.; Chen, L.; Yuan, H.F.; Wang, Y.F.; Yan, X.L.; Xu, Y.C.; Zeng, Q.; et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology 2011, 54, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Wilson, C.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.; Gieling, R.; Chakraborty, J.B.; Fox, C.; et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef] [Green Version]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.Y.; et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013, 58, 139–149. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.I.; Park, O.; Suh, Y.G.; Byun, J.S.; Park, S.Y.; Choi, E.; Kim, J.K.; Ko, H.; Wang, H.; Miller, A.M.; et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes Eugénio, M.; Farooq, M.; Dion, S.; Devisme, C.; Raguenes-Nicol, C.; Piquet-Pellorce, C.; Samson, M.; Dimanche-Boitrel, M.-T.; Le Seyec, J. Hepatocellular carcinoma emergence in diabetic mice with non-alcoholic steatohepatitis depends on diet and is delayed in liver exhibiting an active immune response. Cancers 2020, 12, 1491. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’ s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [CrossRef] [PubMed] [Green Version]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; Group, H.N.I.S. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. [Google Scholar] [CrossRef]
- Llovet, J.M.; Beaugrand, M. Hepatocellular carcinoma: Present status and future prospects. J. Hepatol. 2003, 38, 136–149. [Google Scholar] [CrossRef]
- Tandon, P.; Garcia-Tsao, G. Prognostic indicators in hepatocellular carcinoma: A systematic review of 72 studies. Liver Int. 2009, 29, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Geh, D.; Anstee, Q.M.; Reeves, H.L. NAFLD-associated HCC: Progress and opportunities. J. Hepatocell. Carcinoma 2021, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Gines, P.; Castera, L.; Lammert, F.; Graupera, I.; Serra-Burriel, M.; Allen, A.M.; Wong, V.W.S.; Hartmann, P.; Thiele, M.; Caballeria, L.; et al. Population screening for liver fibrosis: Towards early diagnosis and intervention for chronic liver diseases. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Gailer, R.; Tanwar, S.; Trembling, P.; Parkes, J.; Rodger, A.; Suri, D.; Thorburn, D.; Sennett, K.; Morgan, S.; et al. Prospective evaluation of a primary care referral pathway for patients with non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Berzigotti, A.; Tsochatzis, E.; Boursier, J.; Castera, L.; Cazzagon, N.; Friedrich-Rust, M.; Petta, S.; Thiele, M. EASL Clinical Practice Guidelines on non-invasive tests for evaluation of liver disease severity and prognosis—2021 update. J. Hepatol. 2021, 75, 659–689. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: Expert review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Tapper, E.B.; Castera, L.; Afdhal, N.H. FibroScan (vibration-controlled transient elastography): Where does it stand in the United States practice. Clin. Gastroenterol. Hepatol. 2015, 13, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Choi, K.C.; Wong, G.L.-H.; Zhang, Y.; Chan, H.L.-Y.; Luk, A.O.-Y.; Shu, S.S.-T.; Chan, A.W.-H.; Yeung, M.-W.; Chan, J.C.-N.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: A prospective cohort study. Gut 2016, 65, 1359–1368. [Google Scholar] [CrossRef]
- Pandyarajan, V.; Gish, R.G.; Alkhouri, N.; Noureddin, M. Screening for nonalcoholic fatty liver disease in the primary care clinic. Gastroenterol. Hepatol. 2019, 15, 357. [Google Scholar]
- Sparchez, Z.; Craciun, R.; Caraiani, C.; Horhat, A.; Nenu, I.; Procopet, B.; Sparchez, M.; Stefanescu, H.; Mocan, T. Ultrasound or sectional imaging techniques as screening tools for hepatocellular carcinoma: Fall forward or move forward? J. Clin. Med. 2021, 10, 903. [Google Scholar] [CrossRef]
- Tzartzeva, K.; Obi, J.; Rich, N.E.; Parikh, N.D.; Marrero, J.A.; Yopp, A.; Waljee, A.K.; Singal, A.G. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: A meta-analysis. Gastroenterology 2018, 154, 1706–1718. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Choi, J.-I. Current status of image-based surveillance in hepatocellular carcinoma. Ultrasonography 2021, 40, 45. [Google Scholar] [CrossRef] [PubMed]
- Del Poggio, P.; Olmi, S.; Ciccarese, F.; Di Marco, M.; Rapaccini, G.L.; Benvegnù, L.; Borzio, F.; Farinati, F.; Zoli, M.; Giannini, E.G.; et al. Factors that affect efficacy of ultrasound surveillance for early stage hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2014, 12, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Khalili, K.; Menezes, R.; Kim, T.K.; Yazdi, L.K.; Jang, H.-J.; Sharma, S.; Feld, J.; Sherman, M. The effectiveness of ultrasound surveillance for hepatocellular carcinoma in a Canadian centre and determinants of its success. Can. J. Gastroenterol. Hepatol. 2015, 29, 267–273. [Google Scholar] [CrossRef]
- Nathani, P.; Singal, A.G. Imaging and biomarker approaches to HCC surveillance. Clin. Liver Dis. 2021, 17, 401. [Google Scholar] [CrossRef]
- Nakamura, N.; Hatano, E.; Iguchi, K.; Sato, M.; Kawaguchi, H.; Ohtsu, I.; Sakurai, T.; Aizawa, N.; Iijima, H.; Nishiguchi, S.; et al. Elevated levels of circulating ITIH4 are associated with hepatocellular carcinoma with nonalcoholic fatty liver disease: From pig model to human study. BMC Cancer 2019, 19, 621. [Google Scholar] [CrossRef]
- Best, J.; Bechmann, L.P.; Sowa, J.-P.; Sydor, S.; Dechêne, A.; Pflanz, K.; Bedreli, S.; Schotten, C.; Geier, A.; Berg, T. GALAD score detects early hepatocellular carcinoma in an international cohort of patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2020, 18, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Unić, A.; Derek, L.; Duvnjak, M.; Patrlj, L.; Rakić, M.; Kujundžić, M.; Renjić, V.; Štoković, N.; Dinjar, P.; Jukic, A.; et al. Diagnostic specificity and sensitivity of PIVKAII, GP3, CSTB, SCCA1 and HGF for the diagnosis of hepatocellular carcinoma in patients with alcoholic liver cirrhosis. Ann. Clin. Biochem. 2018, 55, 355–362. [Google Scholar] [CrossRef]
- Yang, J.D.; Addissie, B.D.; Mara, K.C.; Harmsen, W.S.; Dai, J.; Zhang, N.; Wongjarupong, N.; Ali, H.M.; Ali, H.A.; Hassan, F.A.; et al. GALAD score for hepatocellular carcinoma detection in comparison with liver ultrasound and proposal of GALADUS score. Cancer Epidemiol. Biomark. Prev. 2019, 28, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Eddowes, P.J.; Sasso, M.; Allison, M.; Tsochatzis, E.; Anstee, Q.M.; Sheridan, D.; Guha, I.N.; Cobbold, J.F.; Deeks, J.J.; Paradis, V.; et al. Accuracy of FibroScan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2019, 156, 1717–1730. [Google Scholar] [CrossRef] [Green Version]
- Papatheodoridi, M.; Hiriart, J.B.; Lupsor-Platon, M.; Bronte, F.; Boursier, J.; Elshaarawy, O.; Marra, F.; Thiele, M.; Markakis, G.; Payance, A.; et al. Refining the Baveno VI elastography criteria for the definition of compensated advanced chronic liver disease. J. Hepatol. 2021, 74, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Shubrook, J.H.; Adams, L.A.; Pfotenhauer, K.; Wong, V.W.-S.; Wright, E.; Abdelmalek, M.F.; Harrison, S.A.; Loomba, R.; Mantzoros, C.S.; et al. Clinical care pathway for the risk stratification and management of patients with nonalcoholic fatty liver disease. Gastroenterology 2021, 161, 1657–1669. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| HCC Cases Attributable to NAFLD | ||||
|---|---|---|---|---|
| Region | Investigated Population | Year/Period | Prevalence (%) | Reference |
| North America Europe Asia–Pacific Africa | Multicentric study with 42 sites included in 14 countries across the world involving 18,031 patients diagnosed with HCC. | 2012–2015 | 12 10 1–6 12–22 | [20] |
| United States | 4406 HCC patients identified within a healthcare claims database covering 18 million lives yearly and all US census regions. | 2002–2008 | 58 | [19] |
| United States | 26,121 transplanted or waitlisted patients with HCC identified from the Scientific Registry of Transplant Recipients. | 2002 2017 | 2.1 16.2 | [21] |
| United Kingdom | 632 HCC patients consecutively presented at the multidisciplinary team covering North East England, Cumbria and North Yorkshire. | 2000 2010 | <8 34.8 | |
| France | 323 consecutive patients who underwent liver resection due to HCC at two tertiary centers in Paris over a 20-year period. | 1995–1999 2010-2014 | 2.6 19.5 | [22] [23] |
| HCC Arising in the Absence of Cirrhosis | ||||
| Germany United States Japan Germany South Korea | Systematic review with meta-analysis: 19 studies with 168 571 participants and available data about the presence of HCC among patients with/without cirrhosis; 13,345 patients had NAFLD; overall prevalence of HCC in non-cirrhotic NAFLD was 38%. | 2007–2008 2000–2010 2006–2009 1994–2013 2005–2012 | 41.7 26.9 38 13.9 34.3 | [24] |
| Mechanisms | Role in Pathogenesis | |
|---|---|---|
| Single Gene Mutation | PNPLA3, rs738409, p.Ile148Met, chr22 | Increased liver lipid accumulation with a predisposition toward fatty hepatic diseases (NAFLD, NASH, HCC). Influences liver storage of retinol in obese patients, with role in HCC stellate cells to be investigated. |
| TM6SF2, rs58542926, p.E167K, chr19p13.11 | Influences steatosis and advanced fibrosis, independently of DM, obesity or PNPLA3 genotype. Shown in hepatic injury in NAFLD-related HCC, without a clear role in HCC progression. | |
| HFE H63D, rs1799945, chr6p21.3 | Found in non-cirrhotic HCC and led to hepatic inflammation, fibrosis and carcinogensis due to increased parenchymal iron accumulation. | |
| rs641738, chr19q13.42 | Severe liver damage and increased fibrosis risk in NAFLD, with a further investigation regarding HCC progression. | |
| Genetic Instability | DNA amplification of genes involved in oncogenic mechanisms (TERT, VGFA, MET, MYC) | Inducements for NAFLD-related HCC. |
| Oncogene mutations (CTNNB1, AXIN1, ALB, TP53, CDKN2A) | ||
| XPO4 and PDE1B genes | Identified in NAFLD-related HCC; unknown physiological roles in NAFLD-related HCC. | |
| Epigenetic Changes | CHD1 gene | DNA methylation leading to gene silencing; related to DNA damage and repair, lipid and glucose metabolism and fibrosis progression. |
| Dysregulated microRNA Expression | Downregulated liver-specific miR-122 | Reduced expression in NAFLD with negatively regulated hepatic lipogenesis. |
| Other miRNAs’ altered expression (miR-21, miR-29, miR-23, miR-155, miR-221, miR-222, miR-106, miR-93, miR-519) | Major hepatocarcinogenic pathways with several targeting the PTEN protein. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grgurevic, I.; Bozin, T.; Mikus, M.; Kukla, M.; O’Beirne, J. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: From Epidemiology to Diagnostic Approach. Cancers 2021, 13, 5844. https://doi.org/10.3390/cancers13225844
Grgurevic I, Bozin T, Mikus M, Kukla M, O’Beirne J. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: From Epidemiology to Diagnostic Approach. Cancers. 2021; 13(22):5844. https://doi.org/10.3390/cancers13225844
Chicago/Turabian StyleGrgurevic, Ivica, Tonci Bozin, Mislav Mikus, Michal Kukla, and James O’Beirne. 2021. "Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: From Epidemiology to Diagnostic Approach" Cancers 13, no. 22: 5844. https://doi.org/10.3390/cancers13225844