
Melanoma Immunotherapy and Precision Medicine in the Era of Tumor Micro-Tissue Engineering: Where Are We Now and Where Are We Going?


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Melanoma
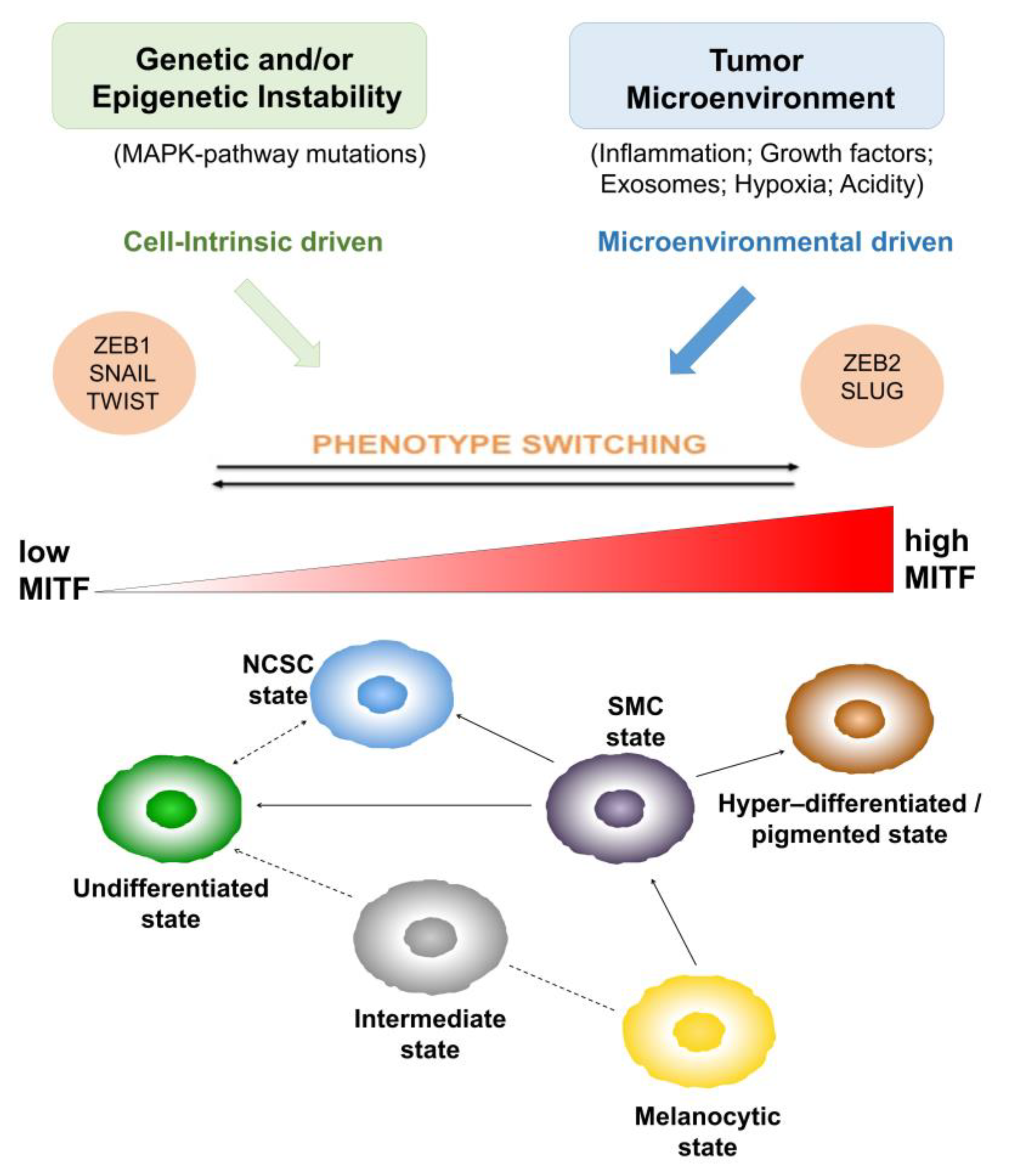
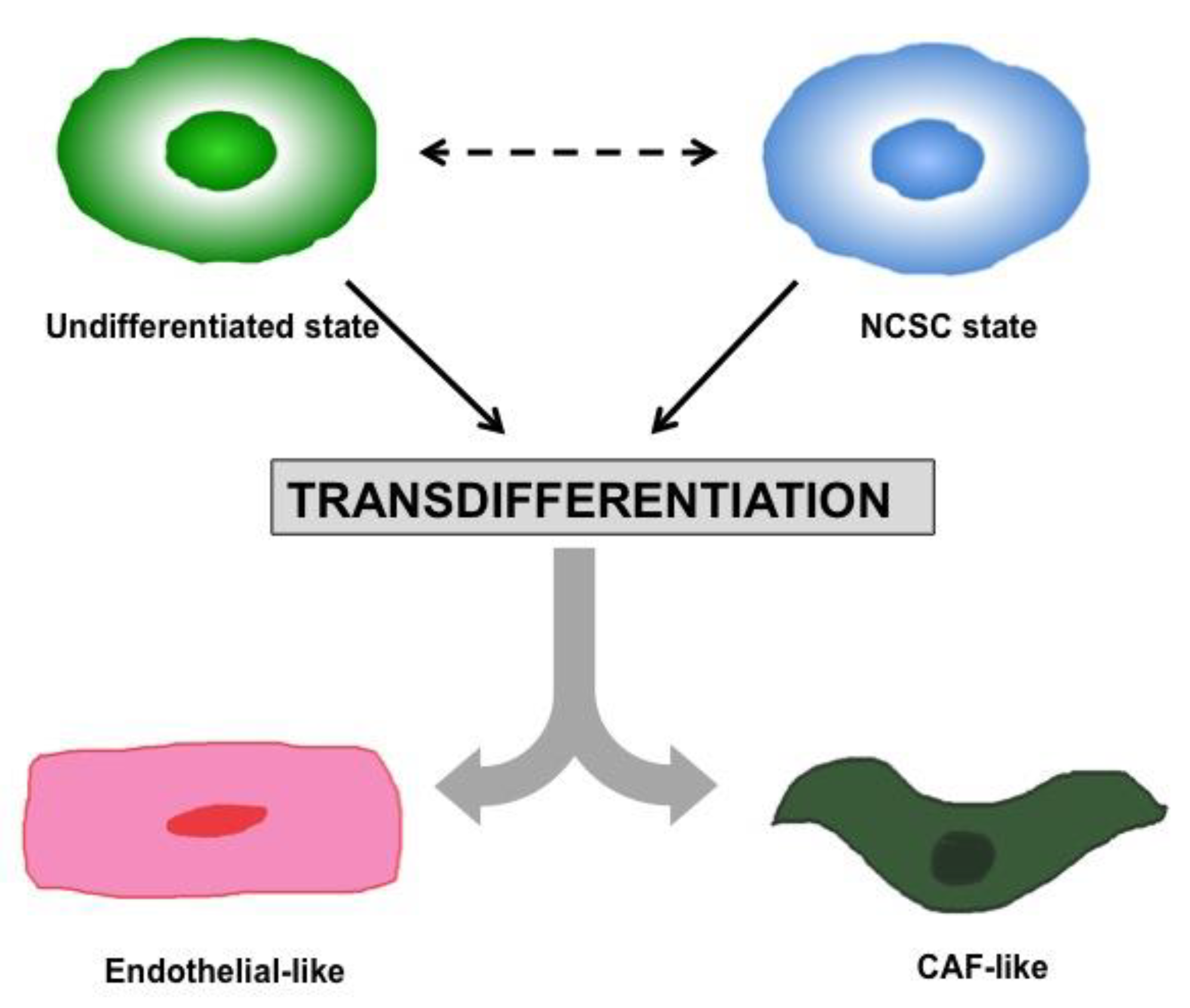
1.2. Heterogeneity and Plasticity: The Most Striking Melanoma Properties
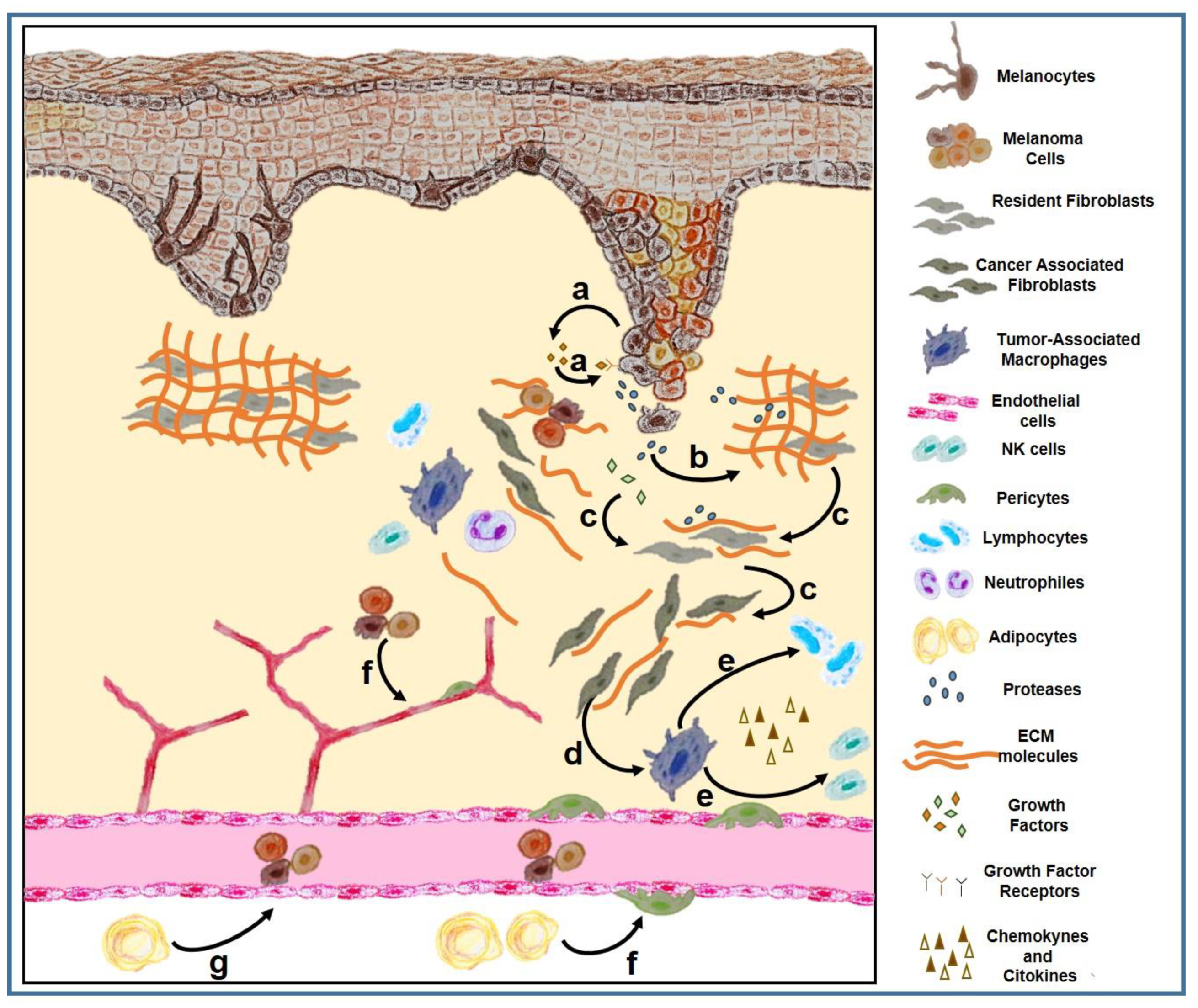
1.3. Tumor Microenvironment
2. Melanoma Modeling
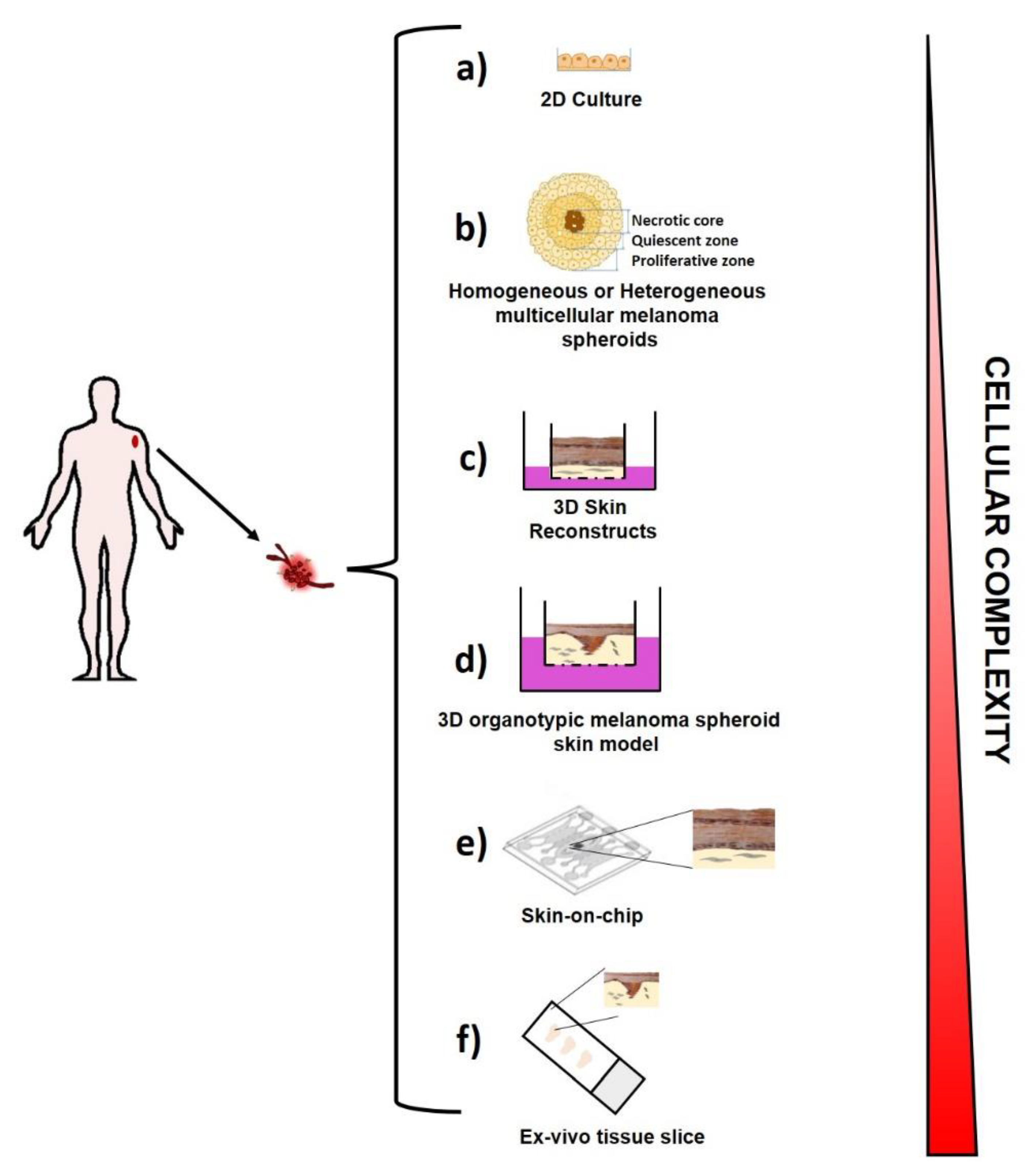
2.1. Ex Vivo Melanoma Models
2.1.1. Two-Dimensional (2D) Melanoma Cell Culture
2.1.2. Three-Dimensional (3D) Melanoma Cell Culture
3. Melanoma Immunotherapy and Precision Medicine: Where We Are Today
4. Melanoma Immunotherapy and Precision Medicine: Where We Are Going in the Tissue Micro-Engineering Era
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base Report on Cutaneous and Noncutaneous Melanoma A Summary of 84,836 Cases from the Past Decade. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 83, 1664–1677. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, L.H.; Larsen, A.-C.; Von Buchwald, C.; Drzewiecki, K.T.; Prause, J.U.; Heegaard, S. Mucosal malignant melanoma-a clinical, oncological, pathological and genetic. APMIS Surv. 2016, 124, 475–486. [Google Scholar] [CrossRef]
- Rodrigues, M.; De Koning, L.; Coupland, S.E.; Jochemsen, A.G.; Marais, R.; Stern, M.-H.; Valente, A.; Barnhill, R.; Cassoux, N.; Evans, A.; et al. cancers Opinion So Close, yet so Far: Discrepancies between Uveal and Other Melanomas. A Position Paper from UM Cure 2020. Cancers 2019, 11, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812. [Google Scholar] [CrossRef]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef]
- Roskoski, R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.Y.; Miller, D.M.; Tsao, H. Somatic driver mutations in melanoma. Cancer 2017, 123, 2104–2117. [Google Scholar] [CrossRef] [Green Version]
- Varrone, F.; Caputo, E. The miRNAs role in melanoma and in its resistance to therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colón-Bolea, P.; García-Gómez, R.; Casar, B. RAC1 Activation as a Potential Therapeutic Option in Metastatic Cutaneous Melanoma. Biomol. 2021, 11, 1554. [Google Scholar] [CrossRef]
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598. [Google Scholar] [CrossRef]
- Haanen, J.B.A.G. Immunotherapy of melanoma. Eur. J. Cancer Suppl. 2013, 11, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choubey, D. Type I interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J. Neuroinflam. 2019, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Coventry, B.J. Therapeutic vaccination immunomodulation: Forming the basis of all cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2019, 7, 2515135519862234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukari, A.; Abdallah, N.; Nagasaka, M. Unleash the power of the mighty T cells-basis of adoptive cellular therapy. Crit. Rev. Oncol. Hematol. 2019, 136, 1–12. [Google Scholar] [CrossRef]
- Chen, C.; Gao, F.-H. Th17 Cells Paradoxical Roles in Melanoma and Potential Application in Immunotherapy. Front. Immunol. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Babacan, N.A.; Eroglu, Z. Treatment Options for Advanced Melanoma After Anti-PD-1 Therapy. Curr. Oncol. Rep. 2020, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Ernstoff, M.S.; Wang, Y.; Menzies, A.M.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.A.; Obeid, M. Autoimmune diseases and immune-checkpoint inhibitors for cancer therapy: Review of the literature and personalized risk-based prevention strategy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 724–744. [Google Scholar] [CrossRef] [PubMed]
- Sadozai, H.; Gruber, T.; Hunger, R.E.; Schenk, M. Recent successes and future directions in immunotherapy of cutaneous melanoma. Front. Immunol. 2017, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, N.; Berx, G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 2014, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Denecker, G.; Vandamme, N.; Akay, Ö.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koen, E.J.; Collier, A.B. Particle-in-cell simulations of a beam driven plasma. Phys. Plasmas 2012, 4, 1420–1428. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Fakultas Psikologi Dan Sosial Budaya Universitas Islam Indonesia Yogyakarta. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197. [Google Scholar] [CrossRef]
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal. Neuron 2003, 38, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Paratore, C.; Goerich, D.E.; Suter, U.; Wegner, M.; Sommer, L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 2001, 128, 3949–3961. [Google Scholar] [CrossRef]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Ii, M.H.W.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [Green Version]
- Madjd, Z.; Erfani, E.; Gheytanchi, E.; Moradi-Lakeh, M.; Shariftabrizi, A.; Asadi-Lari, M. Expression of CD133 cancer stem cell marker in melanoma: A systematic review and meta-analysis. Int. J. Biol. Markers 2016, 31, e118–e125. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Herlyn, M. Melanoma Stem Cells: The Dark Seed of Melanoma. J. Clin. Oncol. 2008, 26, 2890–2894. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Mcallister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2018, 16, 717–727. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O.; Naba, A. Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ruiter, D.; Bogenrieder, T.; Elder, D.; Herlyn, M. Melanoma-stroma interactions: Structural and functional aspects. Lancet. Oncol. 2002, 3, 35–43. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Pilling, D.; Lord, J.M.; Akbar, A.N.; Scheel-Toellner, D.; Salmon, M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001, 22, 199–204. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano: Regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Cancer Cell Perspective Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology 2012, 1, 261–262. [Google Scholar] [CrossRef] [Green Version]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Alizadeh, D.; Katsanis, E.; Larmonier, N. The multifaceted role of Th17 lymphocytes and their associated cytokines in cancer. Clin. Dev. Immunol. 2013, 2013, 957878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Warden, A.R.; Li, Y.; Ding, X. Progress and applications of mass cytometry in sketching immune landscapes. Clin. Transl. Med. 2020, 10, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Maibach, F.; Sadozai, H.; Seyed Jafari, S.M.; Hunger, R.E.; Schenk, M. Tumor-Infiltrating Lymphocytes and Their Prognostic Value in Cutaneous Melanoma. Front. Immunol. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zhang, T.; Michowski, W.; Rebecca, V.W.; Xiao, M.; Ferretti, R.; Suski, J.M.; Bronson, R.T.; Paulo, J.A.; Frederick, D.; et al. Targeting the cyclin-dependent kinase 5 in metastatic melanoma. Proc. Natl. Acad. Sci. USA 2020, 117, 8001–8012. [Google Scholar] [CrossRef]
- Vose, B.M.; Moore, M. Human tumor-infiltrating lymphocytes: A marker of host response. Semin. Hematol. 1985, 22, 27–40. [Google Scholar] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Tumor Angiogenesis. Adv. Cancer Res. 1985, 43, 175–203. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Av Integrins in Angiogenesis and Cancer. Cold Spring Harb. Perspect. Med. 2011, 1, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hida, K.; Ohga, N.; Akiyama, K.; Maishi, N.; Hida, Y. Heterogeneity of tumor endothelial cells. Cancer Sci. 2013, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liang, J.; He, Q.; Wan, Q.; Hou, J.; Lian, K.; Wang, A. The serum biomarker chemerin promotes tumorigenesis and metastasis in oral squamous cell carcinoma. Clin. Sci. 2019, 133, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 421. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Tumour Progression and Recurrence. Nat. Publ. Gr. 2014, 10, 455–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnon, C. Role of the autonomic nervous system in tumorigenesis and metastasis. Mol. Cell. Oncol. 2015, 2, e975643. [Google Scholar] [CrossRef] [Green Version]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.R.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-cancer cell cross-talk: A novel promoter of tumor progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Demir, I.E.; D’Haese, J.G.; Tieftrunk, E.; Kujundzic, K.; Schorn, S.; Xing, B.; Kehl, T.; Friess, H.; Ceyhan, G.O. The neurotrophic factor neurturin contributes toward an aggressive cancer cell phenotype, neuropathic pain and neuronal plasticity in pancreatic cancer. Carcinogenesis 2014, 35, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.M.; Lin, C.; Lin, H.Y.; Chiu, C.M.; Fang, C.W.; Liao, K.F.; Chen, D.R.; Yeh, W.L. Brain-derived neurotrophic factor regulates cell motility in human colon cancer. Endocr. Relat. Cancer 2015, 22, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.R.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, K.; Mohana-Kumaran, N.; Haass, N. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46. [Google Scholar] [CrossRef] [Green Version]
- Park, E.S.; Rabinovsky, R.; Carey, M.; Hennessy, B.T.; Agarwal, R.; Liu, W.; Ju, Z.; Deng, W.; Lu, Y.; Woo, H.G.; et al. Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol. Cancer Ther. 2010, 9, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, E.; Maiorana, L.; Vasta, V.; Pezzino, F.M.; Sunkara, S.; Wynne, K.; Elia, G.; Marincola, F.M.; McCubrey, J.A.; Libra, M.; et al. Characterization of human melanoma cell lines and melanocytes by proteome analysis. Cell Cycle 2011, 10, 2924–2936. [Google Scholar] [CrossRef] [PubMed]
- Andrique, L.; Recher, G.; Alessandri, K.; Pujol, N.; Feyeux, M.; Bon, P.; Cognet, L.; Nassoy, P.; Bikfalvi, A. A model of guided cell self-organization for rapid and spontaneous formation of functional vessels. Sci. Adv. 2019, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34. [Google Scholar] [CrossRef]
- Ramgolam, K.; Lauriol, J.; Lalou, C.; Lauden, L.; Michel, L.; de la Grange, P.; Khatib, A.-M.; Aoudjit, F.; Charron, D.; Alcaide-Loridan, C.; et al. Melanoma spheroids grown under neural crest cell conditions are highly plastic migratory/invasive tumor cells endowed with immunomodulator function. PLoS ONE 2011, 6, e18784. [Google Scholar] [CrossRef]
- Schäfer, M.E.A.; Klicks, J.; Hafner, M.; Rudolf, R. Preparation, Drug Treatment, and Immunohistological Analysis of Tri-Culture Spheroid 3D Melanoma-Like Models. Methods Mol. Biol. 2021, 2265, 173–183. [Google Scholar] [CrossRef]
- Saleh, N.A.; Rode, M.P.; Sierra, J.A.; Silva, A.H.; Miyake, J.A.; Filippin-Monteiro, F.B.; Creczynski-Pasa, T.B. Three-dimensional multicellular cell culture for anti-melanoma drug screening: Focus on tumor microenvironment. Cytotechnology 2021, 73, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4, eaas8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khawar, I.A.; Park, J.K.; Jung, E.S.; Lee, M.A.; Chang, S.; Kuh, H.J. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia 2018, 20, 800–812. [Google Scholar] [CrossRef]
- Riffle, S.; Pandey, R.N.; Albert, M.; Hegde, R.S. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A novel three-dimensional heterotypic spheroid model for the assessment of the activity of cancer immunotherapy agents. Cancer Immunol. Immunother. 2017, 66, 129–140. [Google Scholar] [CrossRef]
- Li, L.; Fukunaga-Kalabis, M.; Herlyn, M. The three-dimensional human skin reconstruct model: A tool to study normal skin and melanoma progression. J. Vis. Exp. 2011, 12, 1–5. [Google Scholar] [CrossRef]
- Gola, M.; Czajkowski, R.; Bajek, A.; Dura, A.; Drewa, T. Melanocyte stem cells: Biology and current aspects. Med. Sci. Monit. 2012, 18, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, C.; Kunisada, M.; Koyanagi-Aoi, M.; Masaki, T.; Takemori, C.; Taniguchi-Ikeda, M.; Aoi, T.; Nishigori, C. Induced pluripotent stem cell-derived melanocyte precursor cells undergoing differentiation into melanocytes. Pigment Cell Melanoma Res. 2019, 32, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Caputo, E.; Miceli, R.; Motti, M.L.; Taté, R.; Fratangelo, F.; Botti, G.; Mozzillo, N.; Carriero, M.V.; Cavalcanti, E.; Palmieri, G.; et al. AurkA inhibitors enhance the effects of B-RAF and MEK inhibitors in melanoma treatment. J. Transl. Med. 2014, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, I.; Kulms, D. A 3D Organotypic Melanoma Spheroid Skin Model. J. Vis. Exp. 2018, 135, e57500. [Google Scholar] [CrossRef] [Green Version]
- Bartfeld, S.; Clevers, H. Organoids as model for infectious diseases: Culture of human and murine stomach organoids and microinjection of helicobacter pylori. J. Vis. Exp. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, J.L.; Young, V.B. A whole new ball game: Stem cell-derived epithelia in the study of host-microbe interactions. Anaerobe 2016, 37, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Doherty, E.L.; Aw, W.Y.; Hickey, A.J.; Polacheck, W.J. Microfluidic and Organ-on-a-Chip Approaches to Investigate Cellular and Microenvironmental Contributions to Cardiovascular Function and Pathology. Front. Bioeng. Biotechnol. 2021, 9, 1–14. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Valencia, L.; Jorcano, J.L.; Velasco, D. Skin-on-a-chip models: General overview and future perspectives. APL Bioeng. 2021, 5, 030901. [Google Scholar] [CrossRef]
- Guenat, O.T.; Berthiaume, F. Incorporating mechanical strain in organs-on-a-chip: Lung and skin. Biomicrofluidics 2018, 12, 042207. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, R.; Wozniak; Edward, P.; Riley; Michael, E.; Charness, M.D. Kidney-on-a-chip: Untapped opportunities HHS Public Access. Physiol. Behav. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver ‘organ on a chip. Exp. Cell Res. 2018, 363, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Meijer, T.G.; Jager, A.; Gent, D.C. Van Ex vivo tumor culture systems for functional drug testing and therapy response prediction-Meijer-2017. Futur. Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [Green Version]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougherara, H.; Mansuet-Lupo, A.; Alifano, M.; Ngô, C.; Damotte, D.; Le Frère-Belda, M.A.; Donnadieu, E.; Peranzoni, E. Real-time imaging of resident T cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front. Immunol. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G. V Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [Green Version]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [Green Version]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers 2020, 12, 1943. [Google Scholar] [CrossRef] [PubMed]
- Sanli, Y.; Leake, J.; Odu, A.; Xi, Y.; Subramaniam, R.M. Tumor Heterogeneity on FDG PET/CT and Immunotherapy: An Imaging Biomarker for Predicting Treatment Response in Patients With Metastatic Melanoma. AJR. Am. J. Roentgenol. 2019, 212, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Rocha Trocoli Drakensjö, I.; Girnita, A. Personalized Medicine in Malignant Melanoma: Towards Patient Tailored Treatment. Front. Oncol. 2018, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Valenti, F.; Falcone, I.; Ungania, S.; Desiderio, F.; Giacomini, P.; Bazzichetto, C.; Conciatori, F.; Gallo, E.; Cognetti, F.; Ciliberto, G.; et al. Precision Medicine and Melanoma: Multi-Omics Approaches to Monitoring the Immunotherapy Response. Int. J. Mol. Sci. 2021, 22, 3837. [Google Scholar] [CrossRef]
- Bourland, J.; Fradette, J.; Auger, F.A. Tissue-engineered 3D melanoma model with blood and lymphatic capillaries for drug development. Sci. Rep. 2018, 8, 13191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Activity: A Case for Synergistic Therapies. Cancer Cell 2018, 31, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Bol, K.F.; Tel, J.; Jolanda, I.; De Vries, M.; Figdor, C.G. Monocyte-and CD34-Derived Dendritic Cells Naturally circulating dendritic cells to vaccinate cancer patients. Oncoimmunology 2013, 2, e23431. [Google Scholar]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5517–5530. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Diao, J.; Mikhailova, A.; Tang, M.; Gu, H.; Zhao, J.; Cattral, M.S. Immunostimulatory conventional dendritic cells evolve into regulatory macrophage-like cells. Blood 2012, 119, 4919–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyfferoen, L.; Brabants, E.; Everaert, C.; De Cabooter, N.; Heyns, K.; Deswarte, K.; Vanheerswynghels, M.; De Prijck, S.; Waegemans, G.; Dullaers, M.; et al. The transcriptome of lung tumor-infiltrating dendritic cells reveals a tumor-supporting phenotype and a microRNA signature with negative impact on clinical outcome. Oncoimmunology 2017, 6, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Shahsafaei, A.; Dorfman, D.M. Germinal-center T-helper-cell markers PD-1 and CXCL13 are both expressed by neoplastic cells in angioimmunoblastic T-cell lymphoma. Am. J. Clin. Pathol. 2009, 131, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Gu-trantien, C.; Sotiriou, C.; Willard-, K.; Gu-trantien, C.; Loi, S.; Garaud, S.; Equeter, C.; Libin, M.; de Wind, A.; Ravoet, M.; et al. CD4 + follicular helper T cell infiltration predicts breast cancer survival Find the latest version: CD4 + follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Investig. 2013, 123, 2873–2892. [Google Scholar] [CrossRef] [PubMed]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2020, 27, 1956–1967. [Google Scholar] [CrossRef]
- Helleday, T. Using personalized immune-humanized xenograft mouse models to predict immune checkpoint responses in malignant melanoma: Potential and hurdles. Ann. Oncol. 2020, 31, 167–168. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W., IV; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, E.; Kuraguchi, M.; Xu, M.; Portell, A.J.; Taus, L.; Diala, I.; Lalani, A.S.; Choi, J.; Chambers, E.S.; Li, S.; et al. Use of Ex Vivo Patient-Derived Tumor Organotypic Spheroids to Identify Combination Therapies for HER2 Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2393–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palechor-Ceron, N.; Krawczyk, E.; Dakic, A.; Simic, V.; Yuan, H.; Blancato, J.; Wang, W.; Hubbard, F.; Zheng, Y.-L.; Dan, H.; et al. Conditional Reprogramming for Patient-Derived Cancer Models and Next-Generation Living Biobanks. Cells 2019, 8, 1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
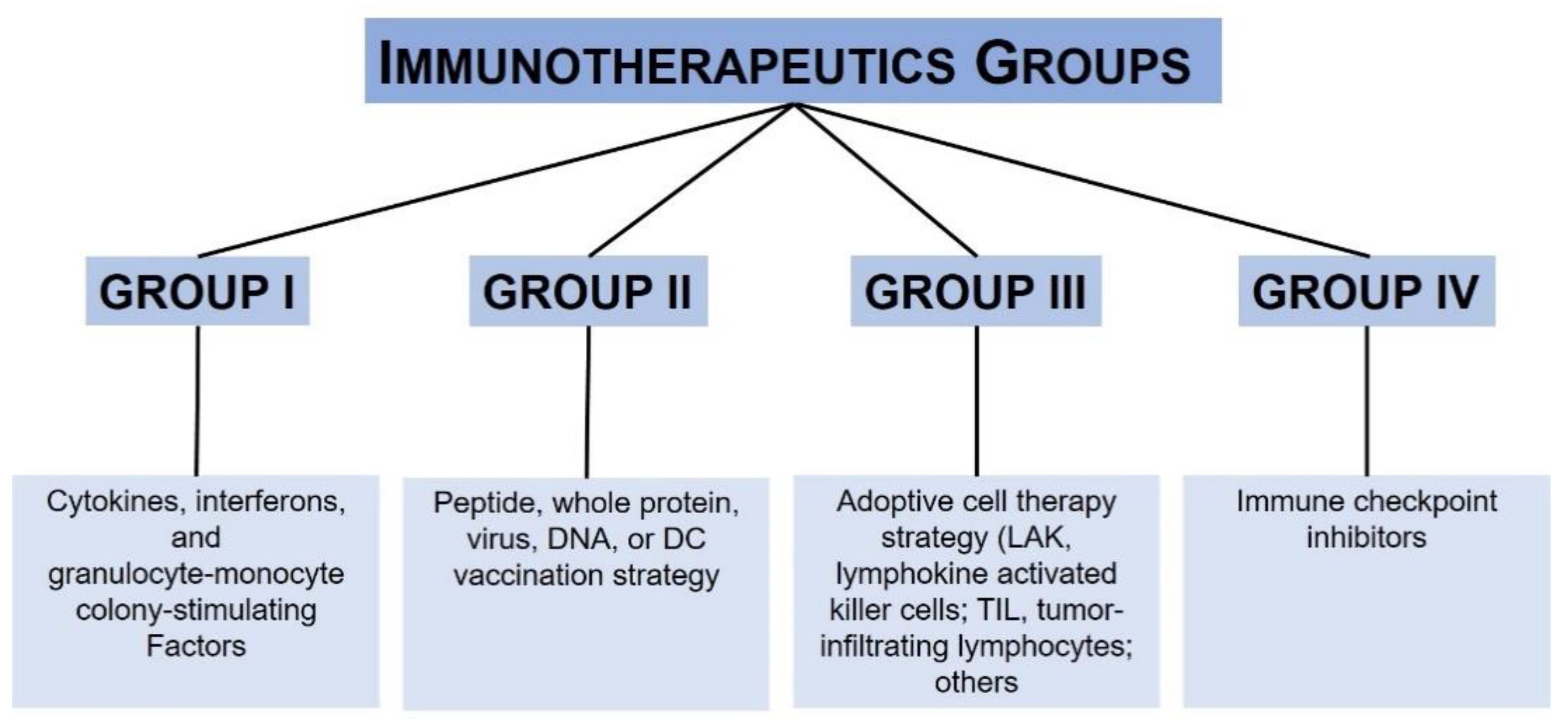{kind=link}
| Gene | Mutated Protein | Frequency (%) * | Drug/ First Approval Date | Target | Note |
|---|---|---|---|---|---|
| B-RAF | V600E V600K V600R | ~60 | Vemurafenib/2011 Dabrafenib/2013 Encorafenib/2018 | BRAFV600E, V600R, V600K kinases | // |
| N-RAS | Q61K Q61R G12D | ~20 | // | // | Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting upstream/downstream NRAS effectors/regulators are in clinical trials |
| MAP2K1/MAP2K2 | E203K E207K | 8 | Trametinib/2013 Cobimetinib/2014 Binimetinib/2017 | MEK1/MEK2 kinases MEK1 Kinase MEK1/MEK2 kinases | AZD8330, TAK-733, GDC-0623 are some of MEK1/2 inhibitors in clinical trials |
| PIK3CA | H1047R E545K | ~5 [12] | // | // | class I PI3K, β-sparing PI3K, PI3Kα inhibitors are in clinical trials |
| RAC1 | P29S | ~4 [13] | Under development [14] | // | Patients carrying RAC1P29S show an increased expression of PD-L1 [15]. Immunotherapy studies by using anti-PD1 or anti PD-L1 antibodies are ongoing |
| Immunotherapy | Drug/First Approval Date | Stage |
|---|---|---|
| PD-1 and PD- L1 inhibitor | Nivolumab (Opdivo®)/2014 Pembrolizumab (Keytruda®)/2014 Atezolizumab (Tecentriq®)/2014 | III |
| CTLA-4 inhibitor | Ipilimumab (Yervoy®)/2011 | III |
| Interferon | Interferon alfa-2b (Intron A®)/2001 Peginterferon alfa-2b (Sylatron®/PEG-Intron®)/2011 | III |
| Interleukin-2 (IL-2, Proleukin) | Aldesleukin (Proleukin®)/1998 | III |
| Oncolytic virus | T-VEC (Imlygic®)/2015 | III–IV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varrone, F.; Mandrich, L.; Caputo, E. Melanoma Immunotherapy and Precision Medicine in the Era of Tumor Micro-Tissue Engineering: Where Are We Now and Where Are We Going? Cancers 2021, 13, 5788. https://doi.org/10.3390/cancers13225788
Varrone F, Mandrich L, Caputo E. Melanoma Immunotherapy and Precision Medicine in the Era of Tumor Micro-Tissue Engineering: Where Are We Now and Where Are We Going? Cancers. 2021; 13(22):5788. https://doi.org/10.3390/cancers13225788
Chicago/Turabian StyleVarrone, Francesca, Luigi Mandrich, and Emilia Caputo. 2021. "Melanoma Immunotherapy and Precision Medicine in the Era of Tumor Micro-Tissue Engineering: Where Are We Now and Where Are We Going?" Cancers 13, no. 22: 5788. https://doi.org/10.3390/cancers13225788