
Oncogenic KRAS: Signaling and Drug Resistance


Abstract
:Simple Summary
Abstract
1. KRAS Protein
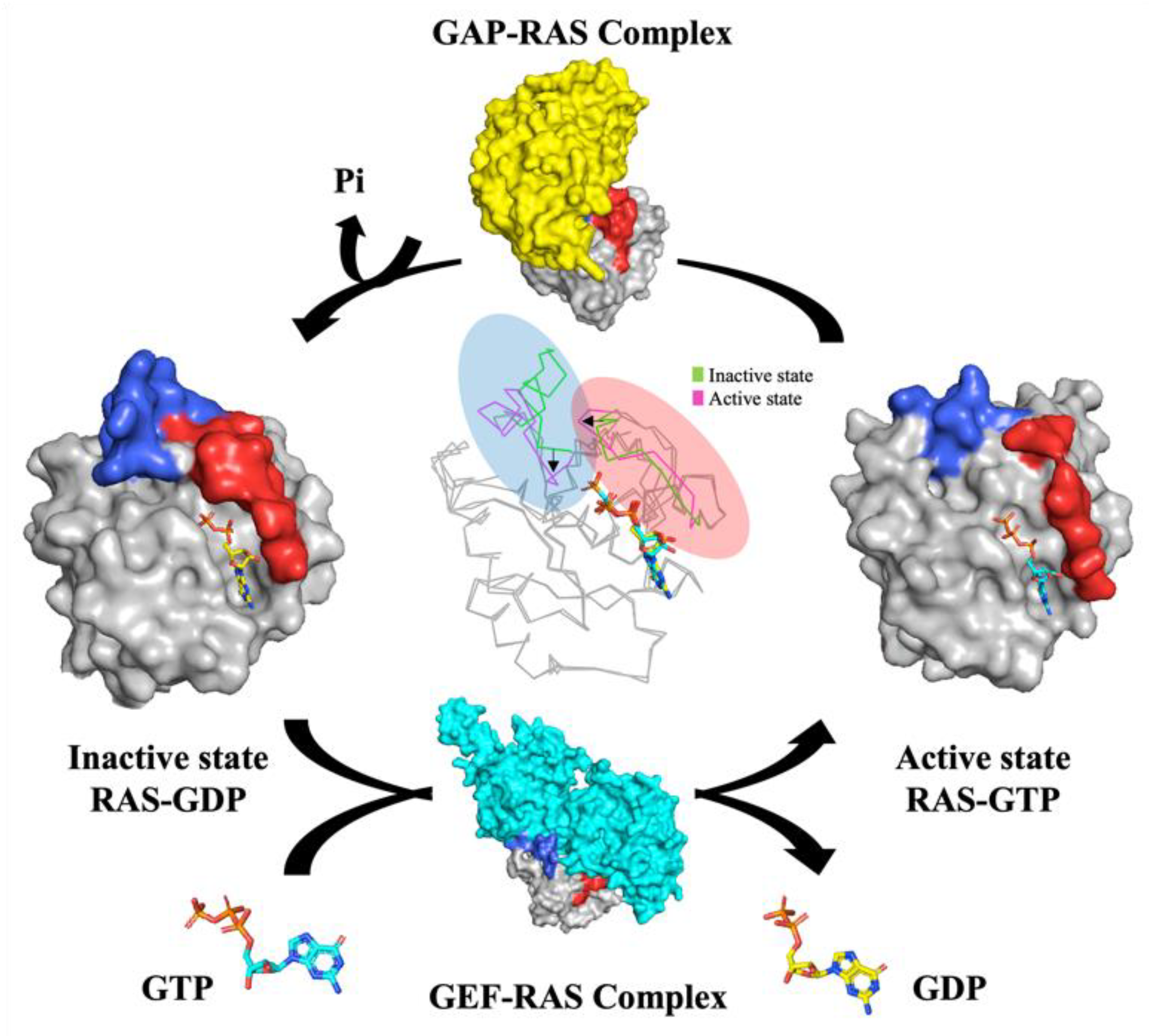
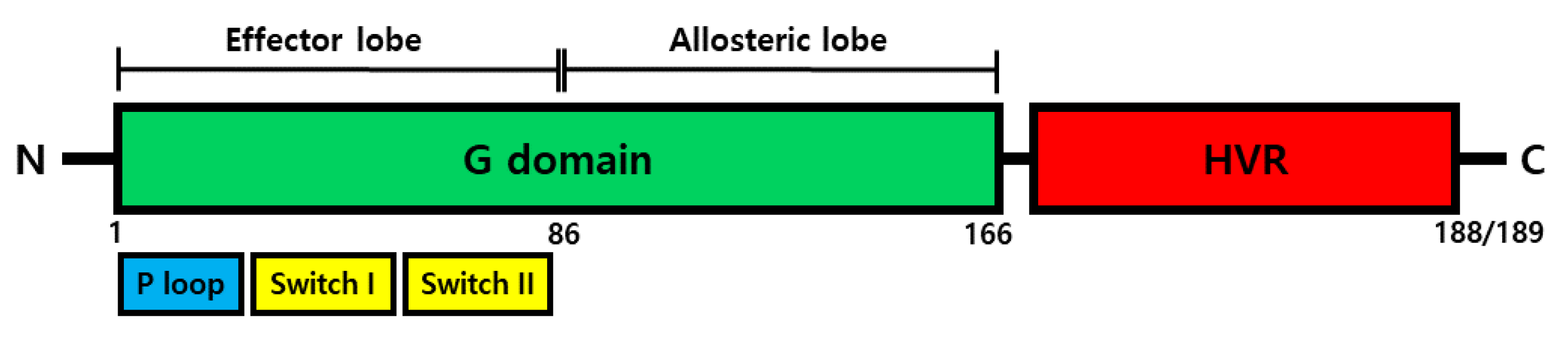
KRAS Structure, Function
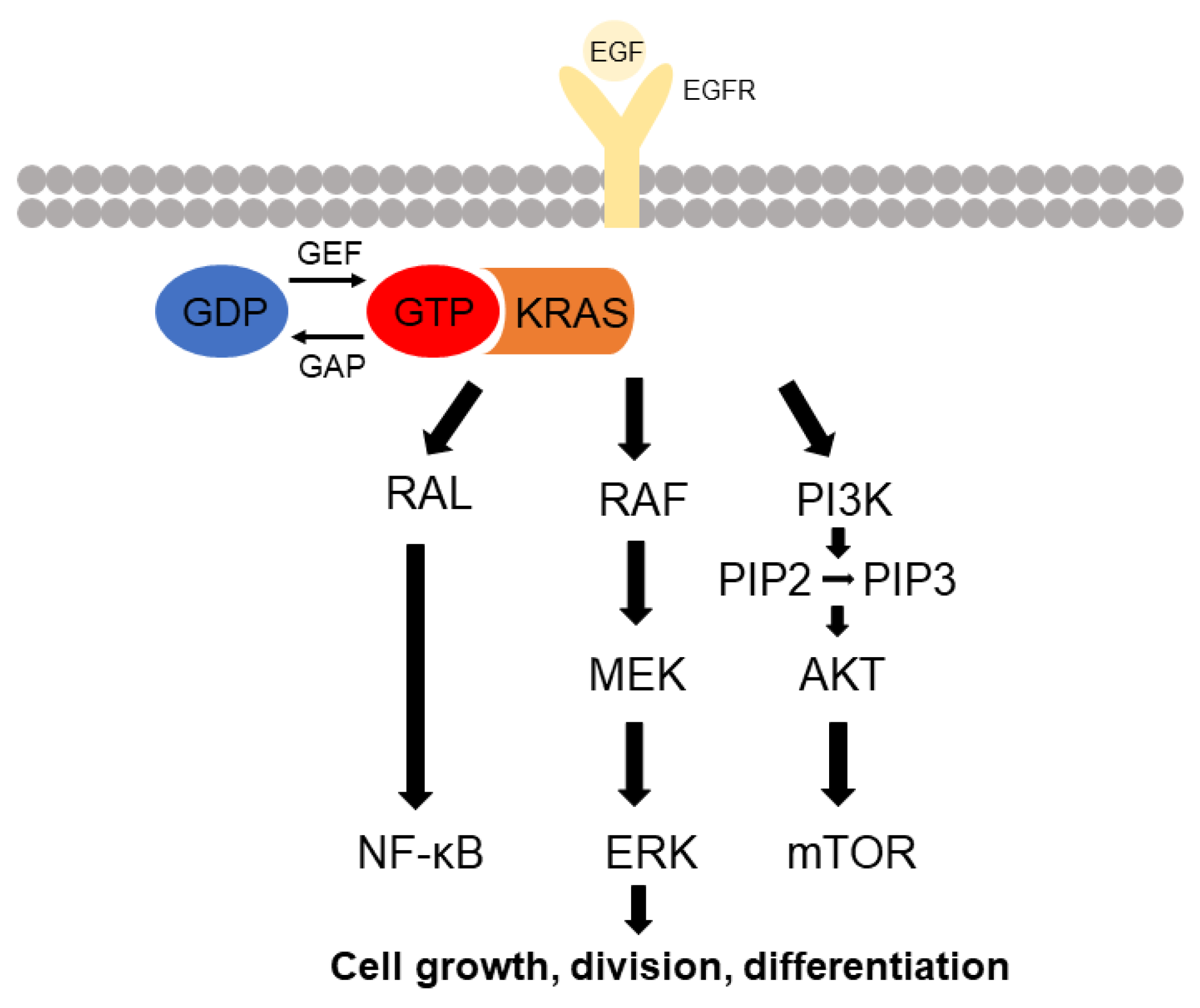
2. KRAS Signaling
2.1. RAF-MEK-ERK Pathway
2.2. PI3K Pathway
2.3. Ral-GEF Protein
3. RAS Inhibitors and Resistance
3.1. Inhibiting the Post-Translation Processing of RAS
3.2. K-RAS Targeted Drug
3.3. Inhibition of RAS Signaling Networks
3.4. KRAS Inhibitors Discovered by Virtual Screening
3.5. Other Inhibitors Targeting RAS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
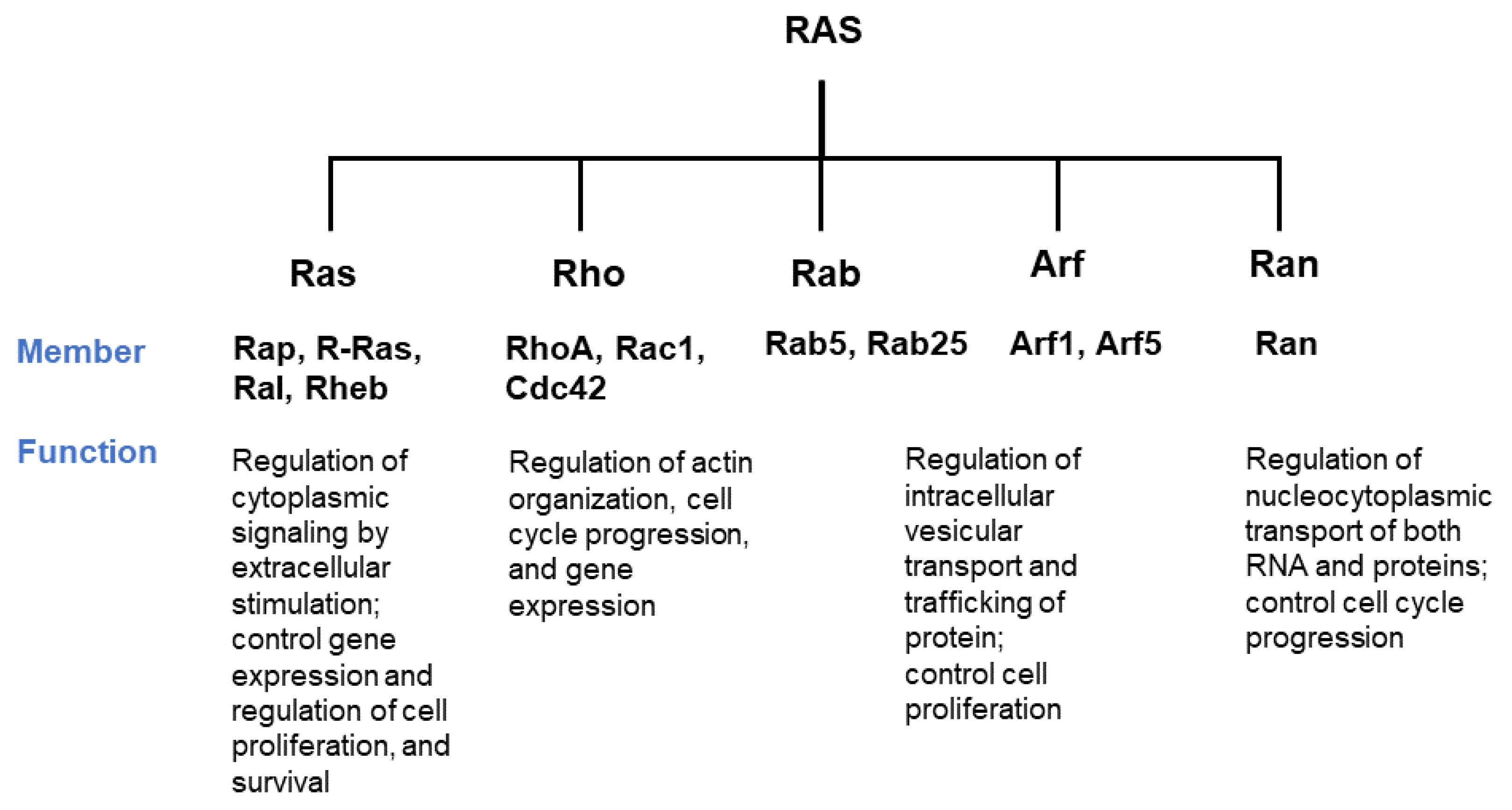
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef] [Green Version]
- Merrick, B.A.; Phadke, D.P.; Bostrom, M.A.; Shah, R.R.; Wright, G.M.; Wang, X.; Gordon, O.; Pelch, K.E.; Auerbach, S.S.; Paules, R.S. Arsenite malignantly transforms human prostate epithelial cells in vitro by gene amplification of mutated KRAS. PLoS ONE 2019, 14, e0215504. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.P.; Capon, D.J.; Smith, D.H.; Chen, E.Y.; Seeburg, P.H.; Goeddel, D.V.; Levinson, A.D. Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature 1983, 304, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, J.E.; Wittinghofer, A. Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: The C-terminal domain of GAP is not sufficient for full activity. Mol. Cell. Biol. 1992, 12, 2050–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.G.; Schaefer, A.; Howard, S.V.; Der, C.J. RAS and RHO family GTPase mutations in cancer: Twin sons of different mothers? Crit. Rev. Biochem. Mol. Biol. 2020, 55, 386–407. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational modifications of RAS proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef]
- Choy, E.; Chiu, V.K.; Silletti, J.; Feoktistov, M.; Morimoto, T.; Michaelson, D.; Ivanov, I.E.; Philips, M.R. Endomembrane trafficking of ras: The CAAX motif targets proteins to the ER and Golgi. Cell 1999, 98, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Abraham, S.J.; Muhamed, I.; Nolet, R.; Yeung, F.; Gaponenko, V. Expression, purification, and characterization of soluble K-Ras4B for structural analysis. Protein Expr. Purif. 2010, 73, 125–131. [Google Scholar] [CrossRef]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS variant signalling. Biochem. Soc. Trans. 2018, 46, 1325. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Mattos, C. The K-Ras, N-Ras, and H-Ras isoforms: Unique conformational preferences and implications for targeting oncogenic mutants. Cold Spring Harb. Perspect. Med. 2018, 8, a031427. [Google Scholar] [CrossRef]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta BBA Biomembr. 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [Green Version]
- Thissen, J.A.; Gross, J.M.; Subramanian, K.; Meyer, T.; Casey, P.J. Prenylation-dependent association of Ki-Ras with microtubules: Evidence for a role in subcellular trafficking. J. Biol. Chem. 1997, 272, 30362–30370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X. Ras activation of the Raf kinase: Tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 2001, 56, 127–155. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The ras/raf/mapk pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warne, P.H.; Vician, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Settleman, J.; Kyriakis, J.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21 ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar] [CrossRef]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef]
- Schlesinger, T.K.; Fanger, G.R.; Yujiri, T.; Johnson, G.L. The tao of MEKK. Front. Biosci 1998, 3, D1181–D1186. [Google Scholar] [CrossRef] [Green Version]
- Arboleda, M.J.; Eberwein, D.; Hibner, B.; Lyons, J.F. Dominant negative mutants of mitogen-activated protein kinase pathway. Methods Enzymol. 2001, 332, 353–367. [Google Scholar] [PubMed]
- Zheng, C.-F.; Guan, K.-L. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J. 1994, 13, 1123–1131. [Google Scholar] [CrossRef]
- Lyons, J.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a novel Raf kinase inhibitor. Endocr.-Relat. Cancer 2001, 8, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Downward, J. Role of RAS in the regulation of PI 3-kinase. Phosphoinositide 3-Kinase Health Dis. 2010, 1, 143–169. [Google Scholar]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.K.; Baker, A.C.; DeBiasi, R.M.; Winckler, W.; LaFramboise, T.; Lin, W.M.; Wang, M.; Feng, W.; Zander, T.; MacConaill, L.E. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 2007, 39, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Simi, L.; Pratesi, N.; Vignoli, M.; Sestini, R.; Cianchi, F.; Valanzano, R.; Nobili, S.; Mini, E.; Pazzagli, M.; Orlando, C. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am. J. Clin. Pathol. 2008, 130, 247–253. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, D.R.; Novick, S.; Kahn, J.M.; Leonardi, P.; Pellicer, A. rsc: A novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene 1997, 14, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.-H.; O’Hayer, K.; Adam, S.J.; Kendall, S.D.; Campbell, P.M.; Der, C.J.; Counter, C.M. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 2006, 16, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Healy, F.M.; Prior, I.A.; MacEwan, D.J. The importance of Ras in drug resistance in cancer. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Kumar, B.; Ahmad, R.; Sharma, S.; Gowrikumar, S.; Primeaux, M.; Rana, S.; Natarajan, A.; Oupicky, D.; Hopkins, C.R.; Dhawan, P. PIK3C3 Inhibition Promotes Sensitivity to Colon Cancer Therapy by Inhibiting Cancer Stem Cells. Cancers 2021, 13, 2168. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, P.; Luo, J.; Wang, J.; Liu, Z.; Wu, W.; Du, Y.; Ye, B.; Wang, D.; He, L. LncRNA HAND2-AS1 promotes liver cancer stem cell self-renewal via BMP signaling. EMBO J. 2019, 38, e101110. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lin, J.; Zhang, H.; Zhu, F.; Xie, R. LncRNA HAND2-AS1 sponging miR-1275 suppresses colorectal cancer progression by upregulating KLF14. Biochem. Biophys. Res. Commun. 2018, 503, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Der, C.J.; Cox, A.D. The Role of Wild Type RAS Isoforms in Cancer; Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 60–69. [Google Scholar]
- Casey, P.J.; Solski, P.A.; Der, C.J.; Buss, J.E. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA 1989, 86, 8323–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.-y. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta BBA Rev. Cancer 2005, 1756, 127–144. [Google Scholar] [CrossRef]
- Bergo, M.O.; Ambroziak, P.; Gregory, C.; George, A.; Otto, J.C.; Kim, E.; Nagase, H.; Casey, P.J.; Balmain, A.; Young, S.G. Absence of the CAAX endoprotease Rce1: Effects on cell growth and transformation. Mol. Cell. Biol. 2002, 22, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter-Vann, A.M.; Baron, R.A.; Wong, W.; dela Cruz, J.; York, J.D.; Gooden, D.M.; Bergo, M.O.; Young, S.G.; Toone, E.J.; Casey, P.J. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4336–4341. [Google Scholar] [CrossRef] [Green Version]
- Boufaied, N.; Wioland, M.-A.; Falardeau, P.; Gourdeau, H. TLN-4601, a novel anticancer agent, inhibits Ras signaling post Ras prenylation and before MEK activation. Anti-Cancer Drugs 2010, 21, 543–552. [Google Scholar] [CrossRef]
- Liu, M.; Sjogren, A.-K.M.; Karlsson, C.; Ibrahim, M.X.; Andersson, K.M.; Olofsson, F.J.; Wahlstrom, A.M.; Dalin, M.; Yu, H.; Chen, Z. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6471–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I. Small molecule inhibition of the KRAS–PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H. BI-3406, a potent and selective SOS1–KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Carver, J.; Dexheimer, T.S.; Hsu, D.; Weng, M.-T.; Smith, J.L.; Guha, R.; Jadhav, A.; Simeonov, A.; Luo, J. A high-throughput assay for small molecule destabilizers of the KRAS oncoprotein. PLoS ONE 2014, 9, e103836. [Google Scholar] [CrossRef]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Ochi, N.; Takigawa, N.; Harada, D.; Yasugi, M.; Ichihara, E.; Hotta, K.; Tabata, M.; Tanimoto, M.; Kiura, K. Src mediates ERK reactivation in gefitinib resistance in non-small cell lung cancer. Exp. Cell Res. 2014, 322, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanis, A.; Ma, H.; Rajkhowa, T.; Ramachandran, A.; Small, D.; Cortes, J.; Levis, M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood J. Am. Soc. Hematol. 2014, 123, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood J. Am. Soc. Hematol. 2017, 129, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Ballard, J.; Blake, J.F.; Bouhana, K.; Brandhuber, B.J.; Briere, D.M.; Burgess, L.E.; Burkard, M.R. Discovery of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C with in vivo activity. ACS Med. Chem. Lett. 2018, 9, 1230–1234. [Google Scholar] [CrossRef]
- Cruz-Nova, P.; Schnoor, M.; Correa-Basurto, J.; Bello, M.; Briseño-Diaz, P.; Rojo-Domínguez, A.; Ortiz-Mendoza, C.M.; Guerrero-Aguirre, J.; García-Vázquez, F.J.; Hernández-Rivas, R. The small organic molecule C19 binds and strengthens the KRAS4b-PDEδ complex and inhibits growth of colorectal cancer cells in vitro and in vivo. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Casique-Aguirre, D.; Briseño-Díaz, P.; García-Gutiérrez, P.; González-de la Rosa, C.H.; Quintero-Barceinas, R.S.; Rojo-Domínguez, A.; Vergara, I.; Medina, L.A.; Correa-Basurto, J.; Bello, M. KRas4B-PDE6δ complex stabilization by small molecules obtained by virtual screening affects Ras signaling in pancreatic cancer. BMC Cancer 2018, 18, 1–16. [Google Scholar] [CrossRef]
- Han, C.W.; Jeong, M.S.; Ha, S.C.; Jang, S.B. A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants. Cancers 2020, 12, 1412. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 1–11. [Google Scholar]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Ryan, M.B.; de la Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, D.; Gmachl, M.; Ramharter, J.; Teh, J.; Fu, S.-C.; Trapani, F.; Kessler, D.; Rumpel, K.; Botesteanu, D.-A.; Ettmayer, P.; et al. BI-3406 and BI 1701963: Potent and selective SOS1:: KRAS inhibitors induce regressions in combination with MEK inhibi-tors or irinotecan. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 22–24 June 2020; Volume 80, p. 1091. [Google Scholar]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Jones, G.G.; Del Río, I.B.; Sari, S.; Sekerim, A.; Young, L.C.; Hartig, N.; Zubiaur, I.A.; El-Bahrawy, M.A.; Hynds, R.E.; Lei, W. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C–Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef]
- Zeng, M.; Lu, J.; Li, L.; Feru, F.; Quan, C.; Gero, T.W.; Ficarro, S.B.; Xiong, Y.; Ambrogio, C.; Paranal, R.M. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem. Biol. 2017, 24, 1005–1016.e3. [Google Scholar] [CrossRef] [Green Version]
- De Vries, G.; Rosas-Plaza, X.; van Vugt, M.A.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef] [PubMed]
- Weykamp, F.; Seidensaal, K.; Rieken, S.; Green, K.; Mende, S.; Zaoui, K.; Freier, K.; Adeberg, S.; Debus, J.; Welte, S.E. Age-dependent hemato-and nephrotoxicity in patients with head and neck cancer receiving chemoradiotherapy with weekly cisplatin. Strahlenther. Onkol. 2020, 196, 515–521. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name | Target | Mode of Inhibition | Ref. |
|---|---|---|---|---|
| Inhibition of post-translational processing | Cysmethynil | ICMT inhibitor | mislocalization of RAS | [51] |
| TLN-4601 | MEK/EKR pathway | disrupts RAS membrane anchorage | [52] | |
| deltarasin | PDEδ | suppresses KRAS localization to endomembrane | [54] | |
| K-RAS targeted drug | ARS-583 | KRAS G12C | inhibits downstream MAPK/PI3K signaling | [55] |
| ARS-1620 | KRAS G12C | anti-tumor activity in subcutaneous xenograft models | [56] | |
| AMG 510 | KRAS G12C | induces pro-inflammatory tumor microenvironment | [57] | |
| MRTX849 | KRAS G12C | tumor regression in KRAS G12C positive cell lines | [58] | |
| BI-3406 | KRAS-mutant of G12 and G13 | interacts with the catalytic domain of SOS, resulting in interference with the interaction with KRAS | [59] | |
| ponatinib and AMG-47a | KRAS G12V | reduces the levels of KRAS G12V proteins | [60] | |
| Inhibition of RAS signaling networks | WZ4002 and MEK inhibitor | EGFR and MEK | effective strategy of drug-resistant cancers and prevents the emergence of drug-resistant clones | [61] |
| gefitinib and Src inhibitor | EGFR and Src | potent strategy to overcome Src-mediated ERK reactivation | [62] | |
| gilteritinib, crenolanib, and midostaurin | FLT3 inhibitor | designed to have a high affinity for the ATP-binding region of the active conformation of FLT3 | [63,64,65] | |
| Discovered by virtual screening | BAY-293 | KRAS-SOS1 interaction | suppresses the RAS-RAF-MEK-ERK pathway and inhibits the activation of RAS in tumor cells | [66] |
| tetrahydro pyridopyrimidines | KRAS G12C | locks KRAS in its inactive GDP-bound form | [67] | |
| C19 | KRAS4B-PDEδ | suppresses the viability and proliferation of colorectal cancer cells | [68] | |
| D14 and C22 | KRAS4B-PDEδ | decreases RAS-GTP activity and ERK and AKT pathways in pancreatic cancer cells | [69] | |
| Other inhibitors | H-REV 107 peptide | KRAS G12V | blocks the activation function of KRAS | [70] |
| cisplatin, carboplatin, and oxaliplatin | widely used to treat several cancers | [71] |
| Primary Tissue | NRAS (%) | KRAS (%) | HRAS (%) |
|---|---|---|---|
| pancreas | 0.77 (4016) | 52.65 (13,817) | 0.11 (3542) |
| colon | 3.87 (16,337) | 32.37 (80,177) | 0.94 (6095) |
| small intestine | 0.45 (443) | 21.34 (1256) | 0.3 (336) |
| lung | 0.91 (17,505) | 14.19 (45,776) | 0.6 (7890) |
| ovary | 1.71 (1932) | 13.03 (6932) | 0.24 (1693) |
| liver | 1.07 (3174) | 3.15 (3614) | 0.24 (2912) |
| kidney | 0.34 (3498) | 0.93 (4423) | 0.25 (3265) |
| skin | 15.36 (15,960) | 2.8 (6833) | 9.42 (7067) |
| endometrium | 3.22 (1648) | 15.45 (4674) | 0.58 (2395) |
| biliary tract | 2.7 (2256) | 18.05 (5518) | 0.56 (1794) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Lee, H.N.; Jeong, M.S.; Jang, S.B. Oncogenic KRAS: Signaling and Drug Resistance. Cancers 2021, 13, 5599. https://doi.org/10.3390/cancers13225599
Kim HJ, Lee HN, Jeong MS, Jang SB. Oncogenic KRAS: Signaling and Drug Resistance. Cancers. 2021; 13(22):5599. https://doi.org/10.3390/cancers13225599
Chicago/Turabian StyleKim, Hyeon Jin, Han Na Lee, Mi Suk Jeong, and Se Bok Jang. 2021. "Oncogenic KRAS: Signaling and Drug Resistance" Cancers 13, no. 22: 5599. https://doi.org/10.3390/cancers13225599