
Mechanical Aspects of Angiogenesis
Abstract
:Simple Summary
Abstract
1. Introduction
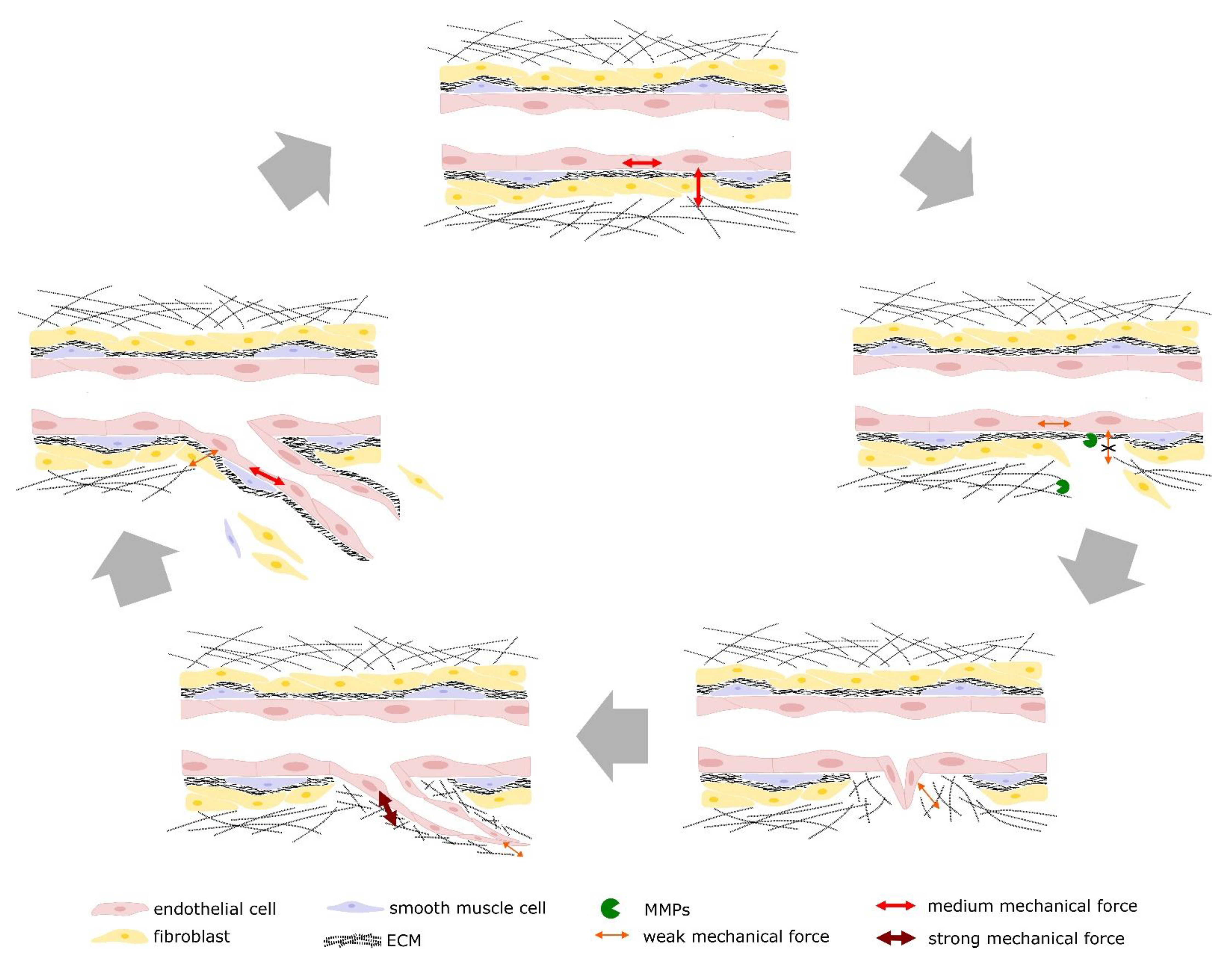
2. Cell-Matrix Contacts
3. Cell Migration
4. Tip/Stalk Cell Selection
5. Vessel Stabilization
6. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Apte, R.S.; Chen, D.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Blanco, R.; Gerhardt, H. VEGF and Notch in Tip and Stalk Cell Selection. Cold Spring Harb. Perspect. Med. 2012, 3, a006569. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical Forces in Tumor Angiogenesis. Biomech. Oncol. 2018, 1092, 91–112. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J.; Kalli, A.C. Force Sensing by Piezo Channels in Cardiovascular Health and Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Boldock, L.; Wittkowske, C.; Perrault, C.M. Microfluidic traction force microscopy to study mechanotransduction in angio-genesis. Microcirculation 2017, 24, 5. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Fujiwara, K.; Pérez, N.G.; Ushio-Fukai, M.; Fisher, A.B. Mechanosignaling in the vasculature: Emerging concepts in sensing, transduction and physiological responses. Am. J. Physiol. Circ. Physiol. 2015, 308, H1451–H1462. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubashkin, M.G.; Cassereau, L.; Bainer, R.; DuFor, C.C.; Yui, Y.; Ou, G.; Paszek, M.J.; Davidson, M.W.; Chen, Y.-Y.; Weaver, V.M. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylino-sitol (3,4,5)-triphosphate. Cancer Res. 2014, 74, 4597–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, M.; Monnot, C.; Muller, L.; Germain, S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin. Cell Dev. Biol. 2018, 89, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.A.; Liliensiek, S.J.; Russell, P.; Nealey, P.F.; Murphy, C.J. Biophysical Cueing and Vascular Endothelial Cell Behavior. Materials 2010, 3, 1620–1639. [Google Scholar] [CrossRef] [Green Version]
- Grant, D.S.; Tashiro, K.-I.; Segui-Real, B.; Yamada, Y.; Martin, G.R.; Kleinman, H.K. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell 1989, 58, 933–943. [Google Scholar] [CrossRef]
- Madri, J.A.; Pratt, B.M.; Yannariello-Brown, J. Matrix-driven cell size change modulates aortic endothelial cell proliferation and sheet migration. Am. J. Pathol. 1988, 132, 18–27. [Google Scholar]
- Fujiwara, H.; Gu, J.; Sekiguchi, K. Rac regulates integrin-mediated endothelial cell adhesion and migration on laminin-8. Exp. Cell Res. 2004, 292, 67–77. [Google Scholar] [CrossRef]
- Kubota, Y.; Kleinman, H.K.; Martin, G.R.; Lawley, T.J. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capil-lary-like structures. J. Cell. Biol. 1988, 107, 1589–1598. [Google Scholar] [CrossRef]
- Rüdiger, D.; Kick, K.; Goychuk, A.; Vollmar, A.M.; Frey, E.; Zahler, S. Cell-Based Strain Remodeling of a Nonfibrous Matrix as an Organizing Principle for Vasculogenesis. Cell Rep. 2020, 32. [Google Scholar] [CrossRef]
- Bonanno, E.; Iurlaro, M.; Madri, J.A.; Nicosia, R.F. Type IV Collagen Modulates Angiogenesis and Neovessel Survival in the Rat Aorta Model. Vitr. Cell. Dev. Biol.-Anim. 2000, 36, 336–340. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Lapiere, C.M.; Nusgens, B.V. In vitro tubulogenesis of endothelial cells by relaxation of the coupling extra-cellular matrix-cytoskeleton. Cardiovasc. Res. 2001, 49, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Senger, D.R. Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 2004, 18, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morpho-genesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottile, J. Regulation of angiogenesis by extracellular matrix. Biochim. et Biophys. Acta (BBA)—Bioenerg. 2004, 1654, 13–22. [Google Scholar] [CrossRef]
- Hynes, R.O. Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 32–40. [Google Scholar] [CrossRef]
- Ingber, D.E. Fibronectin controls capillary endothelial cell growth by modulating cell shape. Proc. Natl. Acad. Sci. USA 1990, 87, 3579–3583. [Google Scholar] [CrossRef] [Green Version]
- Liliensiek, S.J.; Wood, J.A.; Yong, J.; Auerbach, R.; Nealey, P.F.; Murphy, C.J. Modulation of human vascular endothelial cell behaviors by nanotopographic cues. Biomaterials 2010, 31, 5418–5426. [Google Scholar] [CrossRef] [Green Version]
- Reinhart-King, C.; Dembo, M.; Hammer, D.A. The Dynamics and Mechanics of Endothelial Cell Spreading. Biophys. J. 2005, 89, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Califano, J.P.; Reinhart-King, C.A. A Balance of Substrate Mechanics and Matrix Chemistry Regulates Endothelial Cell Net-work Assembly. Cell. Mol. Bioeng. 2008, 1, 122. [Google Scholar] [CrossRef]
- Vailhe, B.; Ronot, X.; Tracqui, P.; Usson, Y.; Tranqui, L. In vitro angiogenesis is modulated by the mechanical properties of fibrin gels and is related to alpha(v)beta3 integ-rin localization. In Vitro Cell Dev. Biol. Anim. 1997, 33, 763–773. [Google Scholar] [CrossRef]
- Shen, C.J.; Raghavan, S.; Xu, Z.; Baranski, J.; Yu, X.; Wozniak, M.A.; Miller, J.; Gupta, M.; Buckbinder, L.; Chen, C.S. Decreased cell adhesion promotes angiogenesis in a Pyk2-dependent manner. Exp. Cell Res. 2011, 317, 1860–1871. [Google Scholar] [CrossRef] [Green Version]
- Mason, B.N.; Starchenko, A.; Williams, R.M.; Bonassar, L.J.; Reinhart-King, C.A. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothe-lial cell behavior. Acta. Biomater. 2013, 9, 4635–4644. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.; Georges, P.C.; Flanagan, L.A.; Marg, B.; Ortiz, M.; Funaki, M.; Zahir, N.; Ming, W.; Weaver, V.M.; Janmey, P.A. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskelet. 2004, 60, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Califano, J.P.; Reinhart-King, C.A. Substrate Stiffness and Cell Area Predict Cellular Traction Stresses in Single Cells and Cells in Contact. Cell. Mol. Bioeng. 2010, 3, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalali, S.; Tafazzoli-Shadpour, M.; Haghighipour, N.; Omidvar, R.; Safshekan, F. Regulation of Endothelial Cell Adherence and Elastic Modulus by Substrate Stiffness. Cell Commun. Adhes. 2015, 22, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.A.; Shah, N.M.; McKee, C.T.; Hughbanks, M.L.; Liliensiek, S.J.; Russell, P.; Murphy, C.J. The role of substratum compliance of hydrogels on vascular endothelial cell behavior. Biomaterials 2011, 32, 5056–5064. [Google Scholar] [CrossRef] [Green Version]
- Winer, J.P.; Oake, S.; Janmey, P.A. Non-Linear Elasticity of Extracellular Matrices Enables Contractile Cells to Communicate Local Position and Orientation. PLoS ONE 2009, 4, e6382. [Google Scholar] [CrossRef] [Green Version]
- Dike, L.E.; Chen, C.S.; Mrksich, M.; Tien, J.; Whitesides, G.M.; Ingber, D.E. Geometric control of switching between growth, apoptosis, and differentiation during angiogenesis using micro-patterned substrates. In Vitro Cell Dev. Biol. Anim. 1999, 35, 441–448. [Google Scholar] [CrossRef]
- Gegenfurtner, F.A.; Jahn, B.; Wagner, H.; Ziegenhain, C.; Enard, W.; Geistlinger, L.; Rädler, J.O.; Vollmar, A.M.; Zahler, S. Micropatterning as a tool to identify regulatory triggers and kinetics of actin-mediated endothelial mech-anosensing. J. Cell Sci. 2018, 131, jcs212886. [Google Scholar] [CrossRef] [Green Version]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial Cell Migration During Angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zouani, O.F.; Rémy, M.; Ayela, C.; Durrieu, M.-C. Geometrical Microfeature Cues for Directing Tubulogenesis of Endothelial Cells. PLoS ONE 2012, 7, e41163. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.L.; Segerer, F.J.; Gegenfurtner, F.A.; Kick, K.; Schreiber, C.; Albert, M.; Vollmar, A.M.; Rädler, J.O.; Zahler, S. Contractility as a global regulator of cellular morphology, velocity, and directionality in low-adhesive fibrillary micro-environments. Biomaterials 2016, 102, 137–147. [Google Scholar] [CrossRef]
- Kick, K.; Nekolla, K.; Rehberg, M.; Vollmar, A.M.; Zahler, S. New View on Endothelial Cell Migration: Switching Modes of Migration Based on Matrix Composition. Arterioscler Thromb. Vasc. Biol. 2016, 36, 2346–2357. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, L.; Underwood, C.J.; Maas, S.; Ellis, B.J.; Kode, T.C.; Hoying, J.B.; Weiss, J.A. Effect of mechanical boundary conditions on orientation of angiogenic microvessels. Cardiovasc. Res. 2008, 78, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.-F.; Yeh, A.T.; Bayless, K.J. Nonlinear optical microscopy reveals invading endothelial cells anisotropically alter three-dimensional collagen matrices. Exp. Cell Res. 2009, 315, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Matsumoto, M.; Patel, S.; Lee, L.; Guan, J.-L.; Li, S. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J. Cell. Physiol. 2004, 203, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.S.; Gardel, M.; Ma, X.; Adelstein, R.; Waterman, C.M. Local Cortical Tension by Myosin II Guides 3D Endothelial Cell Branching. Curr. Biol. 2009, 19, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Bignon, M.; Pichol-Thievend, C.; Hardouin, J.; Malbouyres, M.; Bréchot, N.; Nasciutti, L.; Barret, A.; Teillon, J.; Guillon, E.; Etienne, E.; et al. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 2011, 118, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Reinhart-King, C.A.; Dembo, M.; Hammer, D.A. Cell-Cell Mechanical Communication through Compliant Substrates. Biophys. J. 2008, 95, 6044–6051. [Google Scholar] [CrossRef] [Green Version]
- Saunders, R.L.; Hammer, D.A. Assembly of Human Umbilical Vein Endothelial Cells on Compliant Hydrogels. Cell. Mol. Bioeng. 2010, 3, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2016, 114, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Jannatbabaei, A.; Tafazzoli-Shadpour, M.; Seyedjafari, E. Effects of substrate mechanics on angiogenic capacity and nitric oxide release in human endothelial cells. Ann. N. Y. Acad. Sci. 2020, 1470, 31–43. [Google Scholar] [CrossRef]
- Sun, J.; Jamilpour, N.; Wang, F.-Y.; Wong, P.K. Geometric control of capillary architecture via cell-matrix mechanical interactions. Biomaterials 2014, 35, 3273–3280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, C.; Wang, W.Y.; Zaimi, I.; Jayco, D.K.P.; Baker, B.M. Cell force-mediated matrix reorganization underlies multicellular network assembly. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kniazeva, E.; Putnam, A.J. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am. J. Physiol. Physiol. 2009, 297, C179–C187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kniazeva, E.; Weidling, J.W.; Singh, R.; Botvinick, E.L.; Digman, M.A.; Gratton, E.; Putnam, A.J. Quantification of local matrix deformations and mechanical properties during capillary morphogenesis in 3D. Integr. Biol. 2012, 4, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Francis-Sedlak, M.E.; Moya, M.L.; Huang, J.-J.; Lucas, S.A.; Chandrasekharan, N.; Larson, J.C.; Cheng, M.-H.; Brey, E.M. Collagen glycation alters neovascularization in vitro and in vivo. Microvasc. Res. 2010, 80, 3–9. [Google Scholar] [CrossRef]
- Edgar, L.T.; Underwood, C.J.; Guilkey, J.E.; Hoying, J.B.; Weiss, J.A. Extracellular Matrix Density Regulates the Rate of Neovessel Growth and Branching in Sprouting Angiogenesis. PLoS ONE 2014, 9, e85178. [Google Scholar] [CrossRef] [Green Version]
- Vaeyens, M.-M.; Jorge-Peñas, A.; Fano, J.B.; Steuwe, C.; Heck, T.; Carmeliet, P.; Roeffaers, M.; Van Oosterwyck, H. Matrix deformations around angiogenic sprouts correlate to sprout dynamics and suggest pulling activity. Angiogenesis 2020, 23, 315–324. [Google Scholar] [CrossRef]
- Krishnan, R.; Klumpers, D.D.; Park, C.Y.; Rajendran, K.; Trepat, X.; Van Bezu, J.; Van Hinsbergh, V.W.M.; Carman, C.V.; Brain, J.D.; Fredberg, J.J.; et al. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am. J. Physiol. Physiol. 2011, 300, C146–C154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, N.; Sudo, R.; Ikeda, M.; Tanishita, K. Effects of the mechanical properties of collagen gel on the in vitro formation of microvessel networks by endo-thelial cells. Tissue Eng. 2007, 13, 1443–1453. [Google Scholar] [CrossRef]
- Korff, T.; Augustin, H.G. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J. Cell Sci. 1999, 112. [Google Scholar] [CrossRef]
- Vernon, R.B.; Sage, E.H. Contraction of fibrillar type I collagen by endothelial cells: A study in vitro. J. Cell. Biochem. 1996, 60, 185–197. [Google Scholar] [CrossRef]
- Yung, Y.C.; Chae, J.; Buehler, M.; Hunter, C.P.; Mooney, D.J. Cyclic tensile strain triggers a sequence of autocrine and paracrine signaling to regulate angiogenic sprouting in human vascular cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15279–15284. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Yung, Y.C.; Fischbach, C.; Kong, H.J.; Nakaoka, R.; Mooney, D.J. Mechanical strain regulates endothelial cell patterning in vitro. Tissue Eng. 2007, 13, 207–217. [Google Scholar] [CrossRef]
- Underwood, C.J.; Edgar, L.T.; Hoying, J.B.; Weiss, J.A. Cell-generated traction forces and the resulting matrix deformation modulate microvascular alignment and growth during angiogenesis. Am. J. Physiol. Circ. Physiol. 2014, 307, H152–H164. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, J.; Cheng, A.; Putnam, A.J. Mechanical Strain Controls Endothelial Patterning During Angiogenic Sprouting. Cell. Mol. Bioeng. 2012, 5, 463–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesman, A.; Rosenfeld, D.; Landau, S.; Levenberg, S. Mechanical regulation of vascular network formation in engineered matrices. Adv. Drug Deliv. Rev. 2016, 96, 176–182. [Google Scholar] [CrossRef]
- Vernon, R.B.; Sage, E.H. Between molecules and morphology. Extracellular matrix and creation of vascular form. Am. J. Pathol. 1995, 147, 873–883. [Google Scholar] [PubMed]
- Rongish, B. Relationship of the Extracellular Matrix to Coronary Neovascularization During Development. J. Mol. Cell. Cardiol. 1996, 28, 2203–2215. [Google Scholar] [CrossRef]
- Stenzel, D.; Franco, C.; Estrach, S.; Mettouchi, A.; Sauvaget, D.; Rosewell, I.; Schertel, A.; Armer, H.; Domogatskaya, A.; Rodin, S.; et al. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 2011, 12, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Del Rosso, M.; Fibbi, G.; Pucci, M.; D’Alessio, S.; Del Rosso, A.; Magnelli, L.; Chiarugi, V. Multiple pathways of cell invasion are regulated by multiple families of serine proteases. Clin. Exp. Metastasis 2002, 19, 193–207. [Google Scholar] [CrossRef]
- Laurenzana, A.; Fibbi, G.; Chillà, A.; Margheri, G.; Del Rosso, T.; Rovida, E.; Del Rosso, M.; Margheri, F. Lipid rafts: Integrated platforms for vascular organization offering therapeutic opportunities. Experientia 2015, 72, 1537–1557. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Fibbi, G.; Margheri, F.; Biagioni, A.; Luciani, C.; Del Rosso, M.; Chilla, A. Endothelial Progenitor Cells in Sprouting Angiogenesis: Proteases Pave the Way. Curr. Mol. Med. 2015, 15, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Allen, E.; Apel, I.J.; Gyetko, M.R.; Weiss, S.J. Matrix Metalloproteinases Regulate Neovascularization by Acting as Pericellular Fibrinolysins. Cell 1998, 95, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Santos-Oliveira, P.; Correia, A.; Rodrigues, T.; Ribeiro-Rodrigues, T.M.; Matafome, P.; Rodríguez-Manzaneque, J.C.; Seiça, R.; Girao, H.; Travasso, R.D.M. The Force at the Tip—Modelling Tension and Proliferation in Sprouting Angiogenesis. PLoS Comput. Biol. 2015, 11, e1004436. [Google Scholar] [CrossRef]
- Rivron, N.C.; Vrij, E.J.; Rouwkema, J.; Le Gac, S.; Berg, A.V.D.; Truckenmüller, R.K.; van Blitterswijk, C. Tissue deformation spatially modulates VEGF signaling and angiogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 6886–6891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfahl, H.; Hughes, B.; Alarcon, T.; Maini, P.K.; Lloyd, M.C.; Reuss, M.; Byrne, H.M. 3D hybrid modelling of vascular network formation. J. Theor. Biol. 2017, 414, 254–268. [Google Scholar] [CrossRef]
- Estrach, S.; Cailleteau, L.; Franco, C.A.; Gerhardt, H.; Stefani, C.; Lemichez, E.; Gagnoux-Palacios, L.; Meneguzzi, G.; Mettouchi, A. Laminin-Binding Integrins Induce Dll4 Expression and Notch Signaling in Endothelial Cells. Circ. Res. 2011, 109, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammoto, A.; Connor, K.; Mammoto, T.; Yung, C.W.; Huh, D.; Aderman, C.M.; Mostoslavsky, G.; Smith, L.E.H.; Ingber, D.E. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 2009, 457, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamloo, A.; Mohammadaliha, N.; Heilshorn, S.C.; Bauer, A.L. A Comparative Study of Collagen Matrix Density Effect on Endothelial Sprout Formation Using Experimental and Computational Approaches. Ann. Biomed. Eng. 2015, 44, 929–941. [Google Scholar] [CrossRef]
- Stepanova, V.; Jayaraman, P.-S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.V.; Semina, E.V.; Beloglazova, I.B.; et al. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodo-main Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expres-sion. J. Biol. Chem. 2016, 291, 15029–15045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuss, J.M.; Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adhes. Migr. 2012, 6, 535–540. [Google Scholar] [CrossRef] [Green Version]
- DU, Y.; Herath, S.C.B.; Wang, Q.-G.; Wang, D.-A.; Asada, H.H.; Chen, P.C.Y. Three-Dimensional Characterization of Mechanical Interactions between Endothelial Cells and Extracellular Matrix during Angiogenic Sprouting. Sci. Rep. 2016, 6, 21362. [Google Scholar] [CrossRef] [Green Version]
- Lino, N.; Fiore, L.; Rapacioli, M.; Teruel, L.; Flores, V.; Scicolone, G.; Sánchez, V. uPA-uPAR molecular complex is involved in cell signaling during neuronal migration and neuritogenesis. Dev. Dyn. 2014, 243, 676–689. [Google Scholar] [CrossRef]
- Riahi, R.; Sun, J.; Wang, S.; Long, M.; Zhang, D.D.; Wong, P.K. Notch1–Dll4 signalling and mechanical force regulate leader cell formation during collective cell migration. Nat. Commun. 2015, 6, 6556. [Google Scholar] [CrossRef] [Green Version]
- E Pitulescu, M.; Schmidt, I.; Giaimo, B.D.; Antoine, T.; Berkenfeld, F.; Ferrante, F.; Park, H.; Ehling, M.; Biljes, D.; Rocha, S.F.; et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nature 2017, 19, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.-C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, G.C.; et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. Elife 2018, 7, e31037. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; De Lee, H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Ehling, M.; März, S.; Seebach, J.; Tarbashevich, K.; Sixta, T.; Pitulescu, M.; Werner, A.-C.; Flach, B.; Montanez, E.; et al. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angio-genesis. Nat. Commun. 2017, 8, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauteur, L.; Affolter, M.; Belting, H.G. Distinct and redundant functions of Esama and VE-cadherin during vascular morpho-genesis. Development 2017, 144, 1554–1565. [Google Scholar]
- Kiran, M.S.; I Viji, R.; Kumar, S.V.; A Prabhakaran, A.; Sudhakaran, P.R. Changes in expression of VE-cadherin and MMPs in endothelial cells: Implications for angiogenesis. Vasc. Cell 2011, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Duinen, V.; Zhu, D.; Ramakers, C.; Van Zonneveld, A.J.; Vulto, P.; Hankemeier, T. Perfused 3D angiogenic sprouting in a high-throughput in vitro platform. Angiogenesis 2018, 22, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Takahashi, H.; Pauty, J.; Kobayashi, M.; Kato, K.; Kabara, M.; Kawabed, J.; Matsunaga, Y.T. A 3D in vitro pericyte-supported microvessel model: Visualisation and quantitative characterisation of multistep an-giogenesis. J. Mater Chem. B 2018, 6, 1085–1094. [Google Scholar] [CrossRef]
- Landau, S.; Ben-Shaul, S.; Levenberg, S. Oscillatory Strain Promotes Vessel Stabilization and Alignment through Fibroblast YAP-Mediated Mechanosensitivity. Adv. Sci. 2018, 5, 1800506. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E.; et al. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Dev. Cell 2017, 40, 523–536.e6. [Google Scholar] [CrossRef] [Green Version]
- Fey, T.; Schubert, K.M.; Schneider, H.; Fein, E.; Kleinert, E.; Pohl, U.; Dendorfer, A. Impaired endothelial shear stress induces podosome assembly via VEGF up-regulation. FASEB J. 2016, 30, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Cseh, B.; Fernandez-Sauze, S.; Grall, D.; Schaub, S.; Doma, E.; Van Obberghen-Schilling, E. Autocrine fibronectin directs matrix assembly and crosstalk between cell–matrix and cell–cell adhesion in vascular endothelial cells. J. Cell Sci. 2010, 123, 3989–3999. [Google Scholar] [CrossRef] [Green Version]
- Huynh, J.; Nishimura, N.; Rana, K.; Peloquin, J.; Califano, J.P.; Montague, C.R.; King, M.R.; Schaffer, C.B.; Reinhart-King, C.A. Age-Related Intimal Stiffening Enhances Endothelial Permeability and Leukocyte Transmigration. Sci. Transl. Med. 2011, 3, 112ra122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiger, A.; Liu, F.; Durham, J.; Jagielska, A.; Mahmoodian, R.; Van Vliet, K.; Herman, I. Static mechanical strain induces capillary endothelial cell cycle re-entry and sprouting. Phys. Biol. 2016, 13, 046006. [Google Scholar] [CrossRef] [Green Version]
- Simonavicius, N.; Ashenden, M.; van Weverwijk, A.; Lax, S.; Huso, D.L.; Buckley, C.D.; Huijbers, I.J.; Yarwood, H.; Isacke, C. Pericytes promote selective vessel regression to regulate vascular patterning. Blood 2012, 120, 1516–1527. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.J.; Badu-Nkansah, K.; Hynes, R.O. Endothelium-derived fibronectin regulates neonatal vascular morphogenesis in an autocrine fashion. Angiogenesis 2017, 20, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Forget, A.; Gianni-Barrera, R.; Uccelli, A.; Sarem, M.; Kohler, E.; Fogli, B.; Muraro, M.G.; Bichet, F.; Aumann, K.; Banfi, A.; et al. Mechanically Defined Microenvironment Promotes Stabilization of Microvasculature, Which Correlates with the Enrichment of a Novel Piezo-1(+) Population of Circulating CD11b(+)/CD115(+) Monocytes. Adv. Mater. 2019, 31, e1808050. [Google Scholar] [CrossRef] [PubMed]
- Stratman, A.N.; Malotte, K.M.; Mahan, R.D.; Davis, M.J.; Davis, G.E. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 2009, 114, 5091–5101. [Google Scholar] [CrossRef] [Green Version]
- Gianni-Barrera, R.; Burger, M.; Wolff, T.; Heberer, M.; Schaefer, D.J.; Gürke, L.; Mujagic, E.; Banfi, A. Long-term safety and stability of angiogenesis induced by balanced single-vector co-expression of PDGF-BB and VEGF164 in skeletal muscle. Sci. Rep. 2016, 6, 21546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.; Ng, C.T.; Biniecka, M.; Saber, T.; Taylor, C.; O’Sullivan, J.; Veale, D.J.; Fearon, U. Angiogenesis and blood vessel stability in inflammatory arthritis. Arthritis Rheum. 2010, 62, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.-H.; Skoura, A.; Chae, S.-S.; Cowan, A.E.; Han, D.K.; Proia, R.L.; Hla, T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genome Res. 2004, 18, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Garmy-Susini, B.; Jin, H.; Zhu, Y.; Sung, R.-J.; Hwang, R.; Varner, J. Integrin alpha4beta1-VCAM-1-mediated adhesion between endothelial and mural cells is required for blood vessel maturation. J. Clin. Investig. 2005, 115, 1542–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aplin, A.; Zhu, W.H.; Fogel, E.; Nicosia, R.F. Vascular regression and survival are differentially regulated by MT1-MMP and TIMPs in the aortic ring model of angiogenesis. Am. J. Physiol. Physiol. 2009, 297, C471–C480. [Google Scholar] [CrossRef] [Green Version]
- Staton, C.A.; Reed, M.; Brown, N. A critical analysis of current in vitro and in vivo angiogenesis assays. Int. J. Exp. Pathol. 2009, 90, 195–221. [Google Scholar] [CrossRef]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10, eaao0475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Ferrara, N. Developmental and Pathological Angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. VEGF-A and the Induction of Pathological Angiogenesis. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 251–275. [Google Scholar] [CrossRef]
- Bordeleau, F.; Califano, J.P.; Abril, Y.L.N.; Mason, B.N.; LaValley, D.J.; Shin, S.J.; Weiss, R.S.; Reinhart-King, C.A. Tissue stiffness regulates serine/arginine-rich protein-mediated splicing of the extra domain B-fibronectin isoform in tumors. Proc. Natl. Acad. Sci. USA 2015, 112, 8314–8319. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, K.; Thodeti, C.K.; Dudley, A.C.; Mammoto, A.; Klagsbrun, M.; Ingber, D.E. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11305–11310. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [Green Version]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2017, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Factor | Regulation Process |
|---|---|
| Kind of ECM protein | Different types of proteins have different basic stiffnesses. Different composition and mixing ratios of different types affect the stiffness. ECM proteins elicit specific signaling events. |
| Concentration/density/elasticity (local) | Concentration rise increases stiffness and mechanical barrier. Local changes lead to stiffness gradient. |
| Crosslinking | Increased crosslinking increases stiffness. |
| Intrinsic tension | A tensioned network has an increased fiber stiffness. |
| Degradability | Increased degradability provides reduced resistance to proteolysis resulting in faster reduction of stiffness. |
| Synthesis | Incorporation of additional ECM proteins increases stiffness. |
| Remodeling | Change of architecture increases tension and stiffness of the ECM network. |
| Step of Angiogenesis | Stiffness Modulation | Effect on Angiogenesis | Involved Stiffness Factors |
|---|---|---|---|
| Cell-Matrix contact/adhesion | Increased stiffness increases adhesion | + | Kind of ECM protein Concentration |
| Cell migration | Increased stiffness enhances migration | + | Kind of ECM protein Concentration Intrinsic tension Degradability Remodeling |
| Stiffness gradients determine migration direction | + | ||
| Excessively high stiffness increases the mechanical barrier and reduces migration | − | ||
| Tip/stalk cell selection | Increased stiffness enhances proliferation of stalk cells | +/− (longer sprouts, less branching) | Concentration Synthesis Degradability Remodeling |
| Stiffness gradient promotes tip cell migration | + | ||
| Vessel stabilization | Increased stiffness reduces cell–cell contacts and promotes EC permeability | − | Kind of ECM protein Concentration Crosslinking Synthesis Remodeling |
| Higher stiffness enhances the mural cell recruitment to support EC | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretschmer, M.; Rüdiger, D.; Zahler, S. Mechanical Aspects of Angiogenesis. Cancers 2021, 13, 4987. https://doi.org/10.3390/cancers13194987
Kretschmer M, Rüdiger D, Zahler S. Mechanical Aspects of Angiogenesis. Cancers. 2021; 13(19):4987. https://doi.org/10.3390/cancers13194987
Chicago/Turabian StyleKretschmer, Maibritt, Daniel Rüdiger, and Stefan Zahler. 2021. "Mechanical Aspects of Angiogenesis" Cancers 13, no. 19: 4987. https://doi.org/10.3390/cancers13194987